
SciBase Journals
SciBase Cardiology
ISSN 2691-7785
- Article Type: Case Report
- Volume 1, Issue 1
- Received: Sep 30, 2023
- Accepted: Oct 24, 2023
- Published Online: Oct 31, 2023
Enhancing the Diagnosis of Hypertrophic Cardiomyopathy Diagnosis: A Case of Integrating Cardiac Magnetic Resonance Imaging
Azeem Rathore, DO*; Ali El-Husari, BS; Mohammad Ibrahim, BS; Daanish Khawaja; Niyati Patel, DO; Rafik Jacob, MD
Department of Internal Medicine, The University of Florida College of Medicine, Jacksonville, USA.
*Corresponding Author: Azeem Ratore
Department of Internal Medicine, The University of Florida
College of Medicine, Jacksonville, USA.
Email: azeem.rathore2@jax.ufl.edu
Abstract
Hypertrophic Cardiomyopathy (HCM) is an inherited heart condition characterized by Left Ventricular (LV) wall thickening due to genetic mutations. It can lead to LV outflow tract obstruction, myocardial ischemia, mitral regurgitation, and diastolic dysfunction. A subset, Hypertrophic Obstructive Cardiomyopathy (HOCM), is more prevalent than previously thought. Early HOCM recognition is essential for genetic testing and management, as symptoms are often nonspecific. Diagnosis usually begins with Transthoracic Echocardiography (TTE), which detects LV hypertrophy exceeding 15 mm or lower in familial cases. Cardiac Magnetic Resonance Imaging (CMR) has revolutionized HCM diagnosis, offering precise imaging, tissue characterization, and risk assessment for sudden cardiac events like Sudden Cardiac Death (SCD). This case involves a 30-year-old female with HCM symptoms, initially inconclusively diagnosed via TTE. Subsequent CMR revealed significant LV hypertrophy and HOCMlike features. In summary, CMR is crucial for diagnosing and managing HCM, particularly in cases with inconclusive TTE results. CMR’s advanced imaging aids in precise diagnosis and risk assessment for HCM and complications.
Keywords: Hypertrophic cardiomyopathy; Cardiac MRI; Atrial fibrillation; Transthoracic echocardiogram.
Citation: Rathore A, El-Husari A, Ibrahim M, Khawaja D, Patel N, et al. Enhancing the Diagnosis of Hypertrophic Cardiomyopathy Diagnosis: A Case of Integrating Cardiac Magnetic Resonance Imaging. SciBase Cardiol. 2023; 1(2): 1003.
Introduction
Hypertrophic Cardiomyopathy (HCM) is a commonly inherited heart pathology resulting from a multitude of possible genetic mutations affecting mainly sarcomere proteins [1]. This appears as mutations of the myocardium, showing increased disarray of the myofibrils. HCM is characterized by an increase in Left Ventricular (LV) wall thickness that can lead to Left Ventricular Outflow Tract (LVOT) obstruction, myocardial ischemia, mitral regurgitation, and diastolic dysfunction [1-3]. Hypertrophic Obstructive Cardiomyopathy (HOCM) is a subset of HCM in which significant LVOT obstruction is present and while previously thought of as uncommon, HOCM has a prevalence of 1:200-1:500 [2].
The recognition of HOCM is vital for the proper management of the disease, which includes additional genetic testing, family testing, and medical or surgical management [2-3]. Symptoms of HOCM are often non-specific, including shortness of breath, chest pain, palpitation, syncope, and fatigue [3]. HOCM is commonly identified after symptom onset, heart murmur, abnormal Electrocardiography (ECG), family screening, or imaging unrelated to HOCM. Transthoracic Echocardiography (TTE) is almost always the first imaging modality to diagnose the presence of significant LV hypertrophy that could be suggestive of HCM and HOCM. In most patients, Left Ventricular Hypertrophy (LVH) thickness greater than 15 mm at any site in the cardiac chamber is consistent with the identification of HCM with thickness less than 13-14 mm being diagnostic in cases with confirmed family history of HCM [1]. The process of diagnosing HCM has become significantly more straightforward in recent times, thanks to the advent of Cardiac Magnetic Resonance Imaging (CMR). CMR proves its reliability in confirming the diagnosis, distinguishing HCM from other causes of LVH, and identifying individuals with the highest susceptibility to sudden cardiovascular events, such as Sudden Cardiac Death (SCD). In this context, we present a case of HCM that was diagnosed with the assistance of CMR conducted at our facility.
Case presentation
A 30-year-old female with a past medical history of hypertension and long-standing persistent atrial fibrillation without anticoagulation presented to the emergency department with shortness of breath that started several days prior. She endorsed associated pleuritic pain that she describes as sharp and nonradiating. The patient stated that she has been feeling intermittent shortness of breath for the last few years but that over the past several days her dyspnea became associated with palpitations. She also noted she has been without her amlodipine prescription for the past four days due to financial constraints. Her initial vitals were significant for elevated blood pressure of 217/162 mmHg, 80 beats per minute, respiratory rate of 19 breaths per minute, oxygen saturation of 97% on ambient air, and afebrile. The remaining physical examination was only significant for an irregularly irregular heart rhythm with all other systems unremarkable. Initial laboratory data revealed borderline elevated high-sensitivity cardiac troponin levels (normal values: <14 ng/L) that were serially measured with values of 23 ng/L, 23 ng/L, and 25 ng/L at times zero, one hour, and three hours, respectively, and with a delta of 2 ng/L between zero and three hours. Initial ECG showed atrial fibrillation (AF) without rapid ventricular response along with ST changes in leads V4 -V6 with LVH by the Sokolow-Lyon criteria (Figure 1). Additionally, an elevated D-dimer with a follow-up computed tomography angiography (CTA) revealed a subsegmental pulmonary embolism in the left upper lung lobe. A bedside TTE showed significant LVH along with a preserved ejection fraction, mitral regurgitation, biatrial enlargement, and a dilated inferior vena cava measuring 2.6 cm (normal range: 1.7-2.1 cm) that was not collapsible. Cardiology was subsequently consulted for uncontrolled AF and LVH findings. They recommended initiation of oral anticoagulation, beta-blocker therapy for the hypertensive urgency, and inpatient admission for further work-up of the LVH. On the following day, the patient’s symptoms of shortness of breath and chest pain subsided. A formal TTE was ordered that demonstrated a normal LV size, moderate LVH (23 mm), LV ejection fraction of 60-65%, and an interventricular septum with a phenotypic appearance of HCM though the official interpretation was inconclusive in diagnosis (Figure 2). The patient’s mother later revealed her own history of HCM, and that the patient’s maternal grandmother had died due to “cardiomyopathy” as well. Given this additional context, a CMR was performed for further evaluation that showed septal thickening of 19 mm, lateral wall thickening up to 26 mm consistent with moderate LVH, and an ejection fraction of 39% (Figure 3). Additionally, there was a narrowing of the LVOT with a possible approximation of the anterior mitral valve leaflet with focal interventricular septal hypertrophy. The initial management strategy switched from non-cardioselective beta-blockade therapy to calcium channel therapy with verapamil 80 mg three times daily to decrease the inotropic state of the LV, decrease the gradient across the LVOT, decrease diastolic dysfunction, and increase diastolic filling of the LV by improving diastolic relaxation. The patient’s blood pressure improved with systolic readings ranging between 140 to 150 mmHg. With the resolution of her symptoms, the patient was discharged with expected follow-up with cardiology.
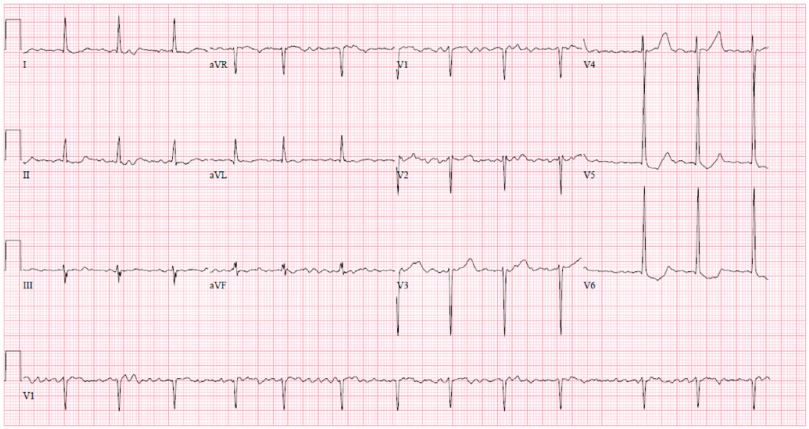
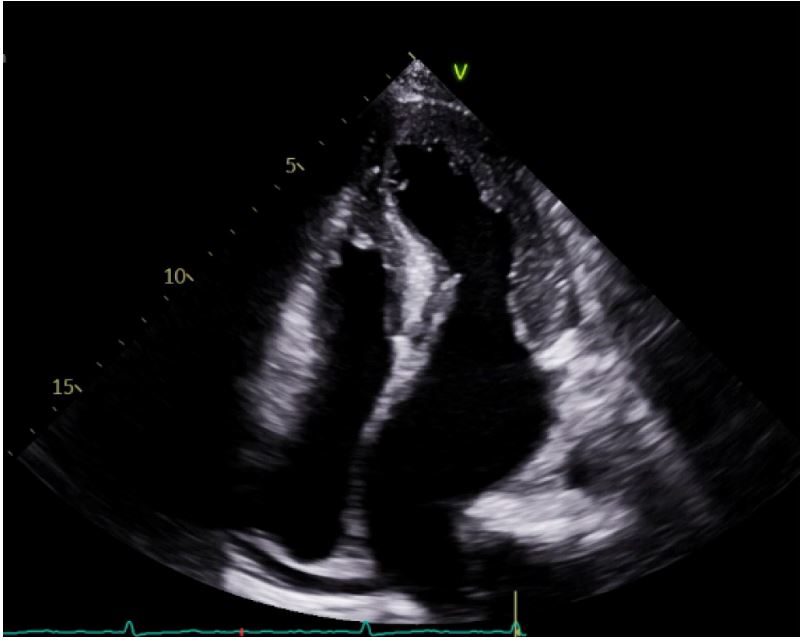
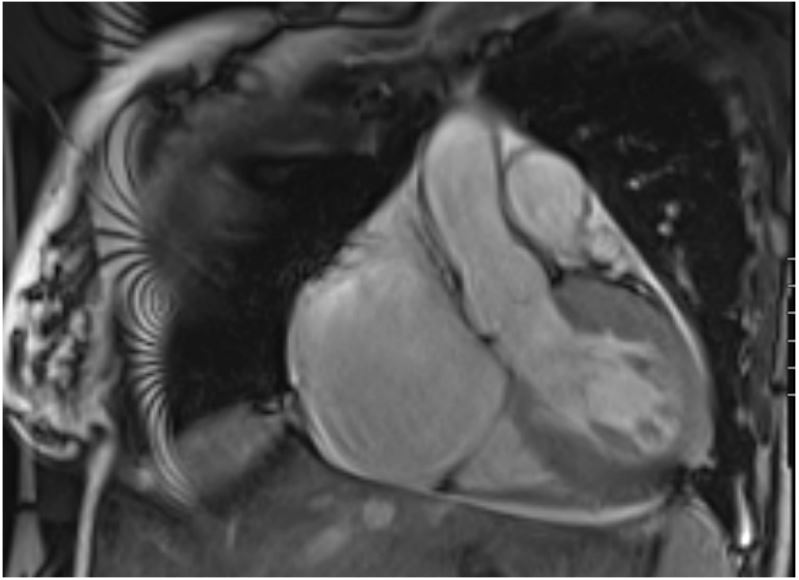
Table 1: Comparison between CMR and TTE [6].
| Aspect | CMR | TTE |
|---|---|---|
| Imaging quality | Excellent soft tissue contrast, high spatial resolution, multiplanar imaging capabilities | Good imaging quality, real-time assessment, lower spatial resolution |
| Acoustic windows | Not dependent on acoustic windows, suitable for patients with poorechocardiographic windows | Dependent on acoustic windows, limited in patients with poorwindows |
| Availability | Less widely available, specialized equipment and expertise required | More widely available, commonly used in routine clinical practice |
| Cost | Generally more expensive | Less expensive |
| Radiation exposure | Uses non-ionizing radiation (magnetic fields and radio waves), no radiation exposure | Does not involve radiation, safe for repeated use |
| Functional assess-ment | Quantitative assessment of cardiac function, tissue characterization,and perfusion | Qualitative assessment of cardiac function, limited tissue characterization |
| Real-time assess-ment | Slower, non-realtime imaging | Real-time imaging |
| Patient comfort | Requires lying still inside an MRI machine for an extended period | Less restrictive, shorter examination time, better tolerated bysome patients |
| Contradictions | Limited in patients with certain metallic implants or severe claustrophobia | Generally safe for most patients, fewer contraindications |
Discussion
HCM is the most common genetic cardiac pathology seen in 0.2-0.5% of the overall population [2]. HCM can affect any portion of the LV but the most widely used diagnostic benchmark is the thickness of the interventricular septum. Unexplained LVH of anywhere greater than 15 mm is diagnostic of HCM [4]. CMR offers superior soft tissue contrast and multiplanar imaging capabilities, making it highly effective for assessing cardiac anatomy and function with excellent spatial resolution [6]. Additionally, CMR does not rely on acoustic windows, allowing for comprehensive evaluation in patients with poor echocardiographic windows, while TTE is more readily available, less expensive, and provides real-time information, making it a valuable initial screening tool for cardiac assessment (Table 1).
Our case report presented a patient with pertinent symptoms of HCM including pleuritic chest pain, shortness of breath, and a strongly suspected family history of hypertrophic cardiomyopathy. Despite the formal TTE revealing significant LVH, CMR was necessary for further evaluation when there is a need for a more detailed assessment of myocardial tissue composition, differentiation of underlying causes of LVH, or evaluation of LVOT obstruction. Additionally, CMR provides valuable information on fibrosis, scar tissue, and precise ventricular function assessment, which can aid in diagnosis and treatment planning for patients with LVH. Indeed, the TTE results showed only mild LVH, and with an increased index of suspicion, the subsequent CMR helped identify more clinically significant LVH and additional findings consistent with HOCM. Our case demonstrates the real-world application of both TTE and CMR and, highlights the utility of CMR when initial TTE findings are seemingly inconclusive.
Conclusion
In conclusion, HCM is an inherited cardiac condition characterized by genetic mutations leading to LV wall thickening and potential complications. Early recognition of HCM and its subset, HOCM, is crucial for effective management. While symptoms of HOCM can be non-specific, TTE is often the initial diagnostic tool. However, the emergence of CMR has greatly improved diagnostic accuracy, offering superior imaging capabilities and risk assessment. In our case, CMR played a pivotal role in diagnosing HCM when TTE results were inconclusive. This emphasizes the importance of a comprehensive diagnostic approach, combining TTE and CMR, to effectively manage HCM and its complications.
References
- Basit H, Brito D, Sharma S. Hypertrophic Cardiomyopathy. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023.
- Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, et al. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022; 79: 372-389.
- Hensley Nadia, Dietrich Jennifer, Nyhan Daniel, Mitter Nanhi, Yee May-Sann, et al. Hypertrophic Cardiomyopathy: A Review. Anesthesia & Analgesia. 2015; 120: 554-569.
- Neupane Nirmal, Rajlawot Kritisha, Koirala Keshika, Phuyal Subash. Cardiac MRI in the Diagnosis and Prognosis of patients with Hypertrophic Cardiomyopathy (HCM) - A Case Report. Nepalese Heart Journal. 2022.
- Maron MS, Lesser JR, Maron BJ. Management implications of massive left ventricular hypertrophy in hypertrophic cardiomyopathy significantly underestimated by echocardiography but identified by cardiovascular magnetic resonance. Am J Cardiol. 2010; 105: 1842-3.
- Hindieh W, Chan R, Rakowski H. Complementary Role of Echocardiography and Cardiac Magnetic Resonance in Hypertrophic Cardiomyopathy. Curr Cardiol Rep. 2017; 19: 81.