
SciBase Journals
SciBase Clinical and Medical Case Reports
ISSN 2995-5874
- Article Type: Case Report
- Volume 2, Issue 1
- Received: Feb 02, 2024
- Accepted: Mar 19, 2024
- Published Online: Mar 26, 2024
Lumbar Contusion in a Young Athlete: Incidental Finding
Sánchez Melús Jorge1*; Mata Crespo; Luz Divina2
1Emergency Department, Ernest Lluch Martin Hospital, Calatayud, Zaragoza, Spain.
2Primary Care, La Muela Health Center, Zaragoza, Spain.
*Corresponding Author: Jorge Sánchez Melús
Emergency Department, Ernest Lluch Martin Hospital, Calatayud, Zaragoza, Spain.
Email: mesanjor@hotmail.com
Abstract
An incidental finding was found in a young football player with no previous disease after a blow in lumbar area. After a proper treatment of a complication (hematuria) and thanks to an image diagnosis, we could diagnose a very common genetic cause of renal failure worldwide but not very usual in Emergency Department and Primary Care Medecine: Autosomal Dominan Polycystic Kidney Disease.
Keywords: Urology; Hematuria; Cysts.
Abbreviations: ADPKD: Autosomal Dominant Polycystic Disease; ARPKD: Autosomal Recessive Polycystic Kidney Disease.
Citation: Jorge SM, Crespo M, Divina L. Lumbar Contusion in a Young Athlete: Incidental Finding. SciBase Clin Med Case Rep. 2024; 2(1): 1018.
Introduction
ADPKD is the most common genetic cause of renal failure worldwide. It is a multisystem and proggresive disease with cyst formation, kidney enlargement and extrarrenal organ involvement (liver, pancreas, spleen). It occurs in all races and is responsable for 6-10% of patients on dialysis in the United States. Cysts may be detected in childhood but clinical manifestations appear between the thrid and fourth decade of life [1]. Furthermore, there is an Autosomal recessive disease, ARPKD, which is rare and it has a much more severe clinical course [2].
Case presentation
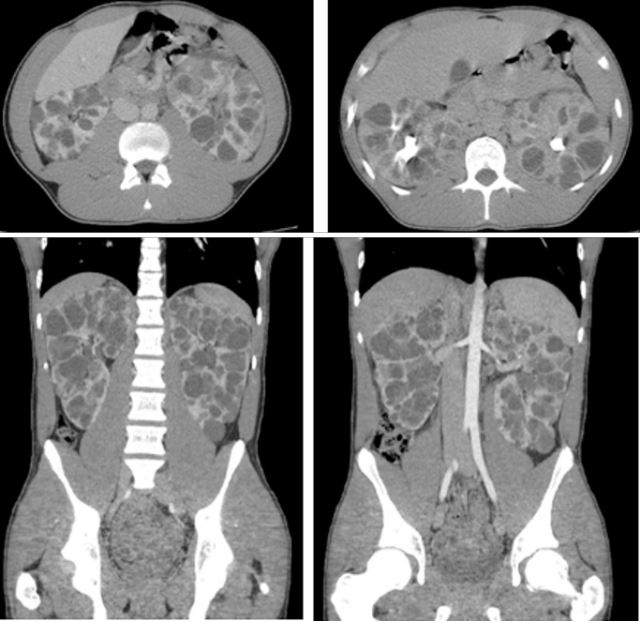
A sixteen year-old male is brought in ambulance to the Emergency Department after a blow in the left lumbar area while he was playing a football match of the local league. No previous disease and no previous surgeries. As his parents explain, after taking a rest, there is no pain relief and something which has scared them is the presence of blood in the urine. No temperature, no other symptoms. First of all, as we recommend with every single patient, we measure vital constants: we are surprised due to his blood pressure (185/111 mmHg), 80 beats/minute, temperatura 36.4o C and SatO2 98%. Also very important to measure (although we know it is very difficult) the pain thanks to the Visual Analog Scale: 3/10 points.Hemodynamically stable, we explore him: he suffers a mechanical non-irradiated lumbar pain with a macroscopic hematuria. No hematomas. The capillary refill is less than one second. The palpation of the abdomen is completely normal, without signs of peritoneal irritation. In this case, it is necessary to extract blood tests with the following results: - Creatinine 1.05 mg/dL; Haemoglobin 13 g/dL and coagulation tests without alterations. Due to these results, we decide request an image test (in this case an abdominal scanner with endovenous contrast): “Bilateral renal polycistic disease, probable automal dominant, with hemorrhagic cysts in both kidneys, more evident on left kidney” (Figure 1).
Discussion
ADPKD involves mutationts in various genes: eighty-five percent of patients have PKD1 (Policystic Kidney Disease 1) mutations (chromosome 16p13.3) and fifteen percent have de PKD2 gene mutation. The most frequent mutation, PKD1, codes for polycystin-1, an integral membrane protein which is present in focal adhesions, primary cilia, tight junctions, desmosomes and adherens junctions. It plays a vital role in cell-to-cell and cellto-matrix interactions [3]. Patients with ADPKD can present a variety of clinical conditions. Kidney fuctioin can remain normal for decades. However, once glomerular function rate starts to decline, renal impairment is usually rapid, with an average loss of 4-5 mL/min/year. Male sex, early age of onset, PKD1 genotype and proteinuria are worse prognostic indicators [1]. As in this case, hypertension is the mos universal and earliest clinical presentation in most patients with ADPKD [4]. Microalbuminuria, proteinuria and hematuria are also mor prevalent in these patients. Episodes of acute flank pain are often seen due to cyst bleeding, infection, stones and (rarely) tumors. Cyst hemorrhage is a frequent complicantion causing gross hematuria when the cyst communicates with the collecting system. Not in this case but prevalence of hepatic cysts increases with age and polycystic liver disease should be suspected when four or more cysts are present in the hepatic parenchyma [5]. About 7 to 36% of patients also have pancreatic cysts, which are more common with PKD2 mutation [6]. When ADPKD is suspected, ultrasound is usually sufficient for asymptomatic patients with normal renal function. Computarized Tomography can help estímate the heighth-adjusted total kidney volumen for risk stratitification of disease progression and may be beneficial for management [1]. As per Renal Association Clinical Practice Guidelines, parents or relatives of individuals with ADPKD should receive education regarding the risk of inheriting ADPKD. Blood pressure should be checked every two years [7].
Ultrasound criteria: Original ravine pkd1 diagnosis criteria [8].
Finally, talking about treatment, we have to manage all complications. Analgesia is required when a patient presents flank pain ore ven a nephorlithiasis, cyst hemorrhage episodes are usually self-limited and it is essential a proper management of blood pressure to reduce cardiovascular mortality and the progression of renal failure. As per the HALT-PKD study, the goal blood pressure range is less than 120-125/80 mmHg, similar to other patients with chronic kidney disease; however, in patients with preserved glomerular function rate is preserved, a lower blood pressure goal (110/75 mmHg) is associated with a decreased incidence of cardiovascular events and a slowe rate of cyst growth [9]. As we used in this patient, angiotensin inhibitors are preferred agents if there is no contraindication. Nephrectomy is only indicated in patients with ADPKD with unbearable abdominal disconfort, non-controlled renal hemorrhage, noncontrolled kidney infections and renal cell carcinoma.
Conclusion
ADPKD is the most common inherited cause of end-stage renal disease worldwide. Although in Primary Care is not very common, we have to be always on alert if there is any family case and the patient has not been studied. It is very important the image diagnosis. In the Emergency Department, we encourage to follow the same order (vital signs, exploration) and try to treat the complication (pain, hemorrhage). Further investigation is needed to find a proper treatment.
Declarations: The authors declare that they have no conflict of interest.
References
- Mahboob M, Rout P, Bokhari SRA. Autosomal Dominant Polycystic Kidney Disease. [Updated 2023 Oct 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; Available from: https://www.ncbi.nlm.nih.gov/books/NBK532934/. 2024.
- Subramanian S, Ahmad T. StatPearls [Internet]. StatPearls Publishing; Treasure Island (FL): Autosomal Recessive Polycystic Kidney Disease. [PubMed]. 2023.
- Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018; 4(1): 50.
- Bell PE, Hossack KF, Gabow PA, Durr JA, Johnson AM, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease. Kidney Int. 1988; 34(5): 683-90.
- Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990; 11(6): 1033-7.
- Kim JA, Blumenfeld JD, Chhabra S, Dutruel SP, Thimmappa ND, Bobb WO, Donahue S, Rennert HE, Tan AY, Giambrone AE, Prince MR. Pancreatic Cysts in Autosomal Dominant Polycystic Kidney Disease: Prevalence and Association with PKD2 Gene Mutations. Radiology. 2016; 280(3): 762-70.
- Dudley J, Winyard P, Marlais M, Cuthell O, Harris T, Chong J, Sayer J, Gale DP, Moore L, Turner K, Burrows S, Sandford R. Clinical practice guideline monitoring children and young people with, or at risk of developing autosomal dominant polycystic kidney disease (ADPKD). BMC Nephrol. 2019; 20(1): 148.
- Petrucci I, Clementi A, Sessa C, Torrisi I, Meola M. Ultrasound and color Doppler applications in chronic kidney disease. J Nephrol. 2018; 31(6): 863-879.
- Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Bae KT, Moore CG, Chapman AB, HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014; 371(24): 2255-66.