
SciBase Journals
SciBase Epidemiology and Public Health
- Article Type: Review Article
- Volume 2, Issue 3
- Received: Sep 05, 2024
- Accepted: Oct 18, 2024
- Published Online: Oct 25, 2024
Theoretical Investigation on Mn(I)-Catalyzed Direct Addition of C(Sp3)−H Bond of 8-Methylquinoline to Aryl Isocyanate Leading to Α-Quinolinyl Amide
Nan Lu†*; Chengxia Miao†
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
*Corresponding Author: Nan Lu
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
Email: lun@sdau.edu.cn
Abstract
Our DFT calculations provide the first theoretical investigation on Mn(I)-catalyzed direct addition of 8-methylquinoline to aryl isocyanate. The reaction of Mn(CO)5Br with Me2Zn forms Mn(CO)5Me, the ligand exchange of which with 8-methylquinoline generates active Me−Mn(I) species after the release of CO. Then Me−Mn(I) undergoes cyclomanganation by elimination of methane giving manganacycle. Subsequently, under the activation of AlCl3, the aryl isocyanate inserts into C−Mn bond producing expanded seven membered manganacycle, the reaction of which with Me2Zn yields methyl manganese species via open at Mn-N. The second ligand exchange regenerates active species for next catalytic cycle and forms precursor of product α-quinolinyl amide via hydrolysis of N. Isocyanate insertion is determined to be rate-limiting. The positive solvation effect is suggested by decreased absolute and activation energies in solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Keywords: Manganese(I); C(sp3)−H bond; Cyclomanganation; Ligand exchange; Isocyanate.
Citation: Lu N, Miao C. Theoretical Investigation on Mn(I)-Catalyzed Direct Addition of C(Sp3)−H Bond of 8-Methylquinoline to Aryl Isocyanate Leading to Α-Quinolinyl Amide. SciBase Epidemiol Public Health. 2024; 2(3): 1028.
Introduction
Due to atom- and step–economy, Grignard-type nucleophilic addition of C−H bonds to polar unsaturated bonds catalyzed by transition-metal has become attracting and potential [1], especially direct addition to polar C-N bond of isocyanate providing amides [2]. The polar C=N bond is easy to integrate with C(sp2)−H bonds with various transition metals, such as Geng’s C−H aminocarbonylation of azobenzenes catalyzed by Re [3], Khan’s spiroannulation of N-acyl ketimines with aryl isothiocyanates through aromatic C−H bond activation using Ru(II) [4], and Li’s aryl/alkenyl C−H aminocarbonylation with acyl azides catalyzed by Co(III) [5]. There are also Ir-catalyzed direct amidation of imidazoles at C-2 leading to imidazole-2-carboxamides of Fukumoto and functionalization of olefinic C−H bonds by aryl-to-vinyl 1,4-Ni migration/reductive coupling sequence of Yang [6,7]. However, only a few examples were reported about addition of C(sp3)−H bonds to isocyanates [8-10].
As efficient transition metal catalysis, Manganese is appealing owing to favorable traits of low cost and low toxicity besides its abundance. Much progress has been made over past few decades in this aspect such as catalytic activation of C−H bonds using Mn(CO)5Br [11]. Liu reported Manganese-catalyzed C(sp2)−H addition to polar unsaturated bonds [12]. Ali summarized recent developments and perspectives in Manganesecatalyzed C−H functionalization driven by weak coordination [13]. Maayuri discovered Manganese-catalyzed hydroarylation of multiple bonds [14]. The notable polar unsaturated bonds are carbonyl compounds, nitriles, and imines. For instance, Liu achieved dimeric Manganese-catalyzed direct nucleophilic addition of C(sp2)−H bonds to inert aldehydes [15]. Das developed one-pot Manganese(I)-catalyzed oxidant-controlled divergent functionalization of 2-arylindazoles [16]. Wang reported Manganese/ NaOPh co-catalyzed C2-selective C−H conjugate addition of indoles to α,β-unsaturated carbonyls [17]. Wang realized redox-neutral C−H acylation of indole with ketene by manganese catalysis [18]. Liang discovered sustainable Manganesecatalyzed C−H activation/hydroarylation of imines [19]. Xu gave access to indole−purine hybrids in Manganese- and Rhenium-catalyzed C−H enaminylation [20].
What we are interested in is the addition to polar C=N bond of isocyanates. The previous progress included Mn(I)-catalyzed C−H aminocarbonylation of heteroarene and [3 + 2] cyclization of ketones and isocyanates via inert C−H activation [21,22]. In this field, the contribution of Wang group is remarkable in particular such as Mn(I)-catalyzed nucleophilic addition of C(sp3)−H bonds to aldehydes yielding α-quinolinyl amide skeleton potential in pharmaceutical compounds [23,24]. Recently, a breakthrough was C(sp3)−H bond aminocarbonylation of 8-methylquinolines with isocyanates catalyzed by Mn(I) [25]. Although 2-(Quinolin-8-yl)-N-(p-tolyl)acetamide was obtained in excellent yields, many problems still puzzled and there was no report about detailed mechanistic study explaining the exact rate-determining step. How the active Me−Mn(I) species is generated via ligand exchange of Mn(CO)5Me with 8-methylquinoline after the release of CO? How methane is eliminated through cyclomanganation giving manganacycle? What’s specific activation process for Lewis acid (AlCl3) in insertion of isocyanate into C−Mn bond? To solve these questions in experiment, an in-depth theoretical study was necessary for this strategy also focusing on the potential ofα-quinolinyl amide.
Computational details
The geometry optimizations were performed at the B3LYP/BSI level with the Gaussian 09 package [26,27]. The mixed basis set of LanL2DZ for Mn, Zn, Br and 6-31G(d) for other non-metal atoms [28-32] was denoted as BSI. Different singlet and multiplet states were clarified with B3LYP and ROB3LYP approaches including Becke’s three-parameter hybrid functional combined with Lee−Yang−Parr correction for correlation [33,34]. The nature of each structure was verified by performing harmonic vibrational frequency calculations. Intrinsic Reaction Coordinate (IRC) calculations were examined to confirm the right connections among key transition-states and corresponding reactants and products. Harmonic frequency calculations were carried out at the B3LYP/BSI level to gain Zero-Point Vibrational Energy (ZPVE) and thermodynamic corrections at 393 K and 1 atm for each structure in 1,4-dioxane. The solvation-corrected free energies were obtained at the B3LYP/6-311++G(d,p) (LanL2DZ for Mn, Zn, Br) level by using Intergral Equation Polarizable Continuum Model (IEFPCM) in Truhlar’s “density” solvation model [35-37] on the B3LYP/BSI-optimized geometries.
As an efficient method of obtaining bond and lone pair of a molecule from modern ab initio wave functions, NBO procedure was performed with Natural Bond Orbital (NBO3.1) to characterize electronic properties and bonding orbital interactions [38,39]. The wave function analysis was provided using Multiwfn_3.7_dev package [40] including research on Frontier Molecular Orbital (FMO) and Mayer Bond Order (MBO).
Results and discussion
The mechanism was explored for Mn(I)-catalyzed direct addition of 8-methylquinoline 1 to aryl isocyanate 2 affording α-quinolinyl amide 3 (Scheme 1). Illustrated by black arrow of Scheme 2, the process is initiated by reaction of Mn(CO)5 with Me2Zn forming intermediate A Mn(CO)5Me. After the release of CO, ligand exchange of A with 8-methylquinoline 1 generates active Me−Mn(I) species B, which undergoes cyclomanganation by elimination of methane to give manganacycle intermediate B-1. Then under the activation of AlCl3, the aryl isocyanate 2 inserts into C−Mn bond of B-1 affording expanded seven membered manganacycle intermediate C. The reaction of C with Me2 gives methyl manganese species D. Ligand exchange of D with 1 forms intermediate E and regenerates active species B for next catalytic cycle. The final product α-quinolinyl amide 3 is produced via hydrolysis of E.
The schematic structures of optimized TSs in Scheme 2 were listed by Figure 1. The activation energy was shown in Table 1 for all steps. Supplementary Table S1, Table S2 provided the relative energies of all stationary points. According to experiment, the Gibbs free energies in 1,4-dioxane solution phase are discussed here.
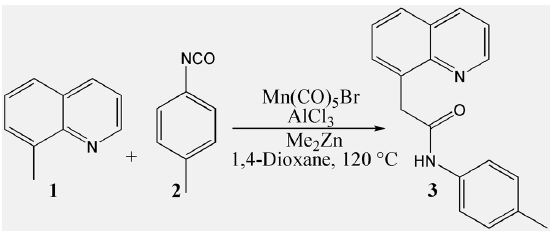

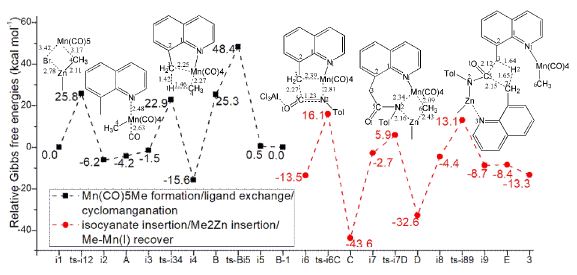
Table 1: Frontal CT scan showing the mass an upper polar tissue mass of the left kidney.
| TS | ΔG≠gas | ΔG≠sol |
|---|---|---|
| ts-i12 | 26.3 | 25.8 |
| ts-i34 | 27.7 | 24.4 |
| ts-Bi5 | 26.4 | 23.1 |
| ts-i6C | 33.9 | 29.6 |
| ts-i7D | 9.1 | 8.6 |
| ts-i89 | 19.8 | 17.5 |
Mn(CO)5Me formation/ligand exchange/cyclomanganation
The reaction of Mn(CO)5Br with Me2Zn proceeds via ts-i12 in step 1 with the activation energy of 25.8 kcal mol−1 relative to the starting point i1 exothermic by -6.2 kcal mol−1 producing i2. The transition vector denotes coordination exchange between Mn and Zn, that is cleavage of Mn···Br, bonding of it to Me and consequent rapture of Zn···Me, linkage of it to Br (3.42, 3.17, 2.11, 2.78 Å) (Figure S1a). After the release of MeZnBr, i2 turns to be another stable intermediate A Mn(CO)5Me with a relative energy of -4.2 kcal mol−1.
Then the participation of 8-methylquinoline 1 with A forms i3, from which the ligand exchange occurs via ts-i34 as step 2 with activation energy of 24.4 kcal mol−1 exothermic by -15.6 kcal mol−1 giving intermediate i4. The transition vector includes one CO ligand leaving from Mn and Mn approaching to N1 of 1 (2.48, 2.63 Å). Once typical Mn-N1 is formed, an active Me−Mn(I) species B is obtained after the removal of CO with a high relative energy of 25.3 kcal mol−1.
The next cyclomanganation is easy to be initiated from B in step 3 via ts-Bi5 with activation energy of 23.1 kcal mol−1 affording i5 endothermic a little by 0.5 kcal mol−1. This process undergoes by elimination of methane to give five membered manganacycle intermediate B-1. According to the transition vector, one hydrogen H1 of 3CH3 is given to 4CH3 ligand of Mn assembling methane molecule 4CH4, which prompts the breaking of Mn···C4 and bonding of Mn···C3 (1.42, 1.46, 2.27, 2.25 Å) (Figure S1b). B-1 involves a structure of five membered manganacycle with same energy of i1.
AlCl3-activated isocyanate insertion/Me2Zn insertion/Me−Mn(I) recover
In step 4, under the activation of AlCl3, the aryl isocyanate 2 inserts into C−Mn bond of B-1 affording expanded seven membered manganacycle intermediate C. The initial complex binding AlCl3, 2 and B-1 is denoted as i6, from which this insertion takes place via ts-i6C with activation energy of 29.6 kcal mol−1 exothermic huge by -43.6 kcal mol−1 . The transition vector corresponds to dissociation of Mn-C3, stretching of C5=N2 from double to single and the resulting linkage of C3···C5, Mn···N2 (2.39, 1.23, 2.27, 2.81 Å). Although the barrier is increased compared with previous three steps, the resultant expanded manganacycle C is rather stable favorable in thermodynamics. Furthermore, the high temperature in experiment ensures the barrier could be overcome completely [25].
Subsequently, the departure of AlCl3 and addition of Me2Zn forms i7, the greatly increased relative energy of which (-2.7 kcal mol−1) suggests the enhanced reactivity of it from that of C. The step 5 happens via ts-i7D with small activation energy of 8.6 kcal mol−1 exothermic by -32.6 kcal mol−1 yielding methyl manganese species D. The transition vector reveals breaking of Mn···N2, Zn···CH3 and concert bonding of Mn-CH3, Zn- N2 (2.34, 2.43, 2.09, 2.16A). Therefore, the seven membered mangana cycle is opened at Mn-N2 binding one Me ligand and Zn isolated.
At last, a second molecule of 1 is added to D generating i8 also with improved reactivity. The second ligand exchange of D with 1 is readily to be initiate via ts-i89 in step 6 with medi activation energy of 17.5 kcal mol−1 exothermic by -8.7 kcal mol−1 delivering intermediate i9 as a binary complexes of active species B and intermediate E. The regenerated B was used for next catalytic cycle. E was taken as precursor of final product α-quinolinyl ethanol 5 via hydrolysis after removal of MeZn and protonation of N2. The detailed atomic motion is illustrated according to the transition vector about donation of H2 from C7 to C3 faciliting the simultaneous broken of C3-C5 and connection of C5-C7 (1.65, 1.64, 2.12, 2.15 Å).
Comparatively, isocyanate insertion activated by AlCl3 of step 4 is determined to be rate-limiting for the whole process. To highlight the idea of feasibility for changes in electron density and not molecular orbital interactions are responsible of the reactivity of organic molecules, quantum chemical tool Multiwfn was applied to analyze of electron density such as MBO results of bonding atoms and contribution of atomic orbital to HOMO of typical TSs (Table S3, Figure S2). These results all confirm the above analysis.
Conclusions
Our DFT calculations provide the first theoretical investigation on Mn(I)-catalyzed direct addition of 8-methylquinoline to aryl isocyanate affording α-quinolinyl amide. The reaction of Mn(CO)5Br with Me2Zn forms intermediate Mn(CO)5Me, the ligand exchange of which with 8-methylquinoline generates active Me−Mn(I) species after the release of CO. Then Me−Mn(I) undergoes cyclomanganation by elimination of methane to give manganacycle intermediate. Subsequently, under the activation of AlCl3, the aryl isocyanate inserts into C−Mn bond producing expanded seven membered manganacycle intermediate, the reaction of which with Me2Zn yields methyl manganese species via open at Mn-N binding Me ligand and Zn isolated. The second ligand exchange with 8-methylquinoline regenerates active species for next catalytic cycle and forms precursor of final product via hydrolysis of N. Comparatively, isocyanate insertion is determined to be rate-limiting for the whole process. The positive solvation effect is suggested by decreased absolute and activation energies in 1,4-dioxane solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Declarations
Author contributions: Conceptualization, Nan Lu; Methodology, Nan Lu; Software, Nan Lu; Validation, Nan Lu; Formal Analysis, Nan Lu; Investigation, Nan Lu; Resources, Nan Lu; Data Curation, Nan Lu; Writing-Original Draft Preparation, Nan Lu; Writing-Review & Editing, Nan Lu; Visualization, Nan Lu; Supervision, Chengxia Miao; Project Administration, Chengxia Miao; Funding Acquisition, Chengxia Miao. All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by National Natural Science Foundation of China (21972079) and Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, P.R. China.
Conflict of interest: The authors declare no conflict of interest.
Electronic supplementary material: Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 393 K.].
References
- Hummel JR, Boerth J. A.; Ellman, J. A. Transition-Metal-Catalyzed C-H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev. 2017; 117: 9163-9227.
- Kumar H, Kumar P, Narasimhan B, Ramasamy K, Mani V, et al. Synthesis, in vitro antimicrobial, antiproliferative, and QSAR studies of N-(substituted phenyl)-2/4-(1H-indol-3-ylazo)-benzamides. Med. Chem. Res. 2013; 22: 1957-1971.
- Geng X, Wang C. Rhenium-catalyzed C-H Aminocarbonylation of Azobenzenes with Isocyanates. Org. Biomol. Chem. 2015; 13: 7619-7623.
- Khan ZA, Singh VK. Synthesis of Spiroisoindolinones via Ru(II)-Catalyzed Spiroannulation of N-Acyl Ketimines with Aryl Isocyanates/Isothio-cyanates through Aromatic C-H Bond Activation. J. Org. Chem. 2023; 88: 17438-17449.
- Li J, Ackermann L. Cobalt (III)-Catalyzed Aryl and Alkenyl C-H Aminocarbonylation with Isocyanates and Acyl Azides. Angew. Chem., Int. Ed. 2015; 54: 8551-8554.
- Fukumoto Y, Shiratani M, Noguchi H, Chatani N. Iridium-Catalyzed Direct Amidation of Imidazoles at the C-2 Position with Isocyanates in the Presence of Hydrosilanes Leading to Imidazole-2-Carboxamides. Synthesis. 2021; 53: 3011-3018.
- Yang J, Gui Z, He Y, Zhu S. Functionalization of Olefinic C-H Bonds by an Aryl-to-Vinyl 1,4-Nickel Migration/Reductive Coupling Sequence. Angew. Chem., Int. Ed. 2023; 62: e202304713.
- Zhao H, Zhou X, Li B, Liu X, Guo N, et al. Rhodium(III)-Catalyzed C(sp 3 )-H Bond Aminocarbonylation with Isocyanates. J. Org. Chem. 2018; 83: 4153-4159.
- Tortajada A, Correia JTM, Serrano E, Monleón A, Tampieri A, et al. Ligand-Controlled Regiodivergent Catalytic Amidation of Unactivated Secondary Alkyl Bromides. ACS Catal. 2021; 11: 10223-10227.
- Kawasaki T, Yamazaki K, Tomono R, Ishida N, Murakami M. Photoinduced Carbamoylation of C(sp3)-H Bonds with Isocyanates. Chem. Lett. 2021; 50: 1684-1687.
- Aneeja T, Neetha M, Afsina CMA, Anilkumar G. Recent advances and perspectives in manganese-catalyzed C-H activation. Catal. Sci. Technol. 2021; 11: 444-458.
- Liu T, Wang C. Manganese-Catalyzed C(sp2)-H Addition to Polar Unsaturated Bonds. Synlett. 2021; 32: 1323-1329.
- Ali S, Rani A, Khan S. Manganese-catalyzed C-H functionalizations driven via weak coordination: Recent developments and perspectives. Tetrahedron Lett. 2022; 97: 153749.
- Maayuri R, Gandeepan P. Manganese-catalyzed hydroarylation of multiple bonds. Org. Biomol. Chem. 2023; 21: 441-464.
- Liu H, Peng J, Li B, Wang B. Dimeric Manganese-Catalyzed Direct Nucleophilic Addition of C(sp2)-H Bonds to Inert Aldehydes. J. Org. Chem. 2022; 87: 14487-14495.
- Das KK, Ghosh AK, Hajra A. One-Pot Manganese(I)-Catalyzed Oxidant-Controlled Divergent Functionalization of 2-Arylindazoles. Chem. - Eur. J. 2024; 30: e202302849.
- Wang Z, Wang C. Manganese/NaOPh co-catalyzed C2-selective C-H conjugate addition of indoles to α,β-unsaturated carbonyls. Green Synth. Catal. 2021; 2: 66-69.
- Wang C, Zhang Q. Redox-neutral C-H acylation of indole with ketene by manganese catalysis. Green Synth. Catal. 2022; 3: 287-290.
- Liang YF, Massignan L, Ackermann L. Sustainable Manganese-Catalyzed C-H Activation/Hydroarylation of Imines. Chem Cat Chem. 2018; 10: 2768-2772.
- Xu Z, Wang Y, Zheng Y, Huang Z, Ackermann L, et al. Manganese- and Rhenium-catalyzed C-H Enaminylation: Expedient Access to Novel Indole-purine Hybrids with Anti-tumor Bioactivities. Org. Chem. Front. 2020; 7: 3709-3714.
- Liu W, Bang J, Zhang Y, Ackermann L. Manganese(I)-Catalyzed C-H Aminocarbonylation of Heteroarenes. Angew. Chem., Int. Ed. 2015; 54: 14137-14140.
- Huo J, Yang Y, Wang C. Manganese-Catalyzed [3 + 2] Cyclization of Ketones and Isocyanates via Inert C-H Activation. Org. Lett. 2021; 23: 3384-3388.
- Liu H, Tang T, Li B, Wang B. Manganese(I)-catalyzed Nucleophilic Addition of C(sp3)-H Bonds to Aldehydes. Chem. Commun. 2024; 60: 5066-5069.
- Sham HL, Konradi AW, Hom RK, Probst GD, Bowers S, et al. PCT International Application. WO Patent. WO0913102010.
- Liu H, Yu Z, Li B, Wang B. Manganese(I)-Catalyzed Direct Addition of C(sp3)-H Bonds to Aryl Isocyanates. J. Org. Chem. 2024. https://doi.org/10.1021/acs.joc.4c01581
- Frisch MJ, Trucks GW, Schlegel HB. et al. Gaussian 09 (Revision B.01), Gaussian, Inc., Wallingford, CT. 2010.
- Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations-potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985; 82: 270-283.
- Lv H, Han F, Wang N, Lu N, Song Z, et al. Ionic Liquid Catalyzed C-C Bond Formation for the Synthesis of Polysubstituted Olefins. Eur. J. Org. Chem. 2022; e202201222.
- Zhuang H, Lu N, Ji N, Han F, Miao C. Bu4NHSO4‐Catalyzed Direct N‐Allylation of Pyrazole and its Derivatives with Allylic Alcohols in Water: A Metal‐free, Recyclable and Sustainable System. Advanced Synthesis & Catalysis. 2021; 363: 5461-5472.
- Lu N, Lan X, Miao C, Qian P. Theoretical investigation on transformation of Cr(II) to Cr(V) complexes bearing tetra-NHC and group transfer reactivity. Int. J. Quantum Chem. 2020; 120: e26340.
- Lu N, Liang H, Qian P, Lan X, Miao C. Theoretical investigation on the mechanism and enantioselectivity of organocatalytic asymmetric Povarov reactions of anilines and aldehydes. Int. J. Quantum Chem. 2020; 120: e26574.
- Lu N, Wang Y. Alloy and Media Effects on the Ethanol Partial Oxidation Catalyzed by Bimetallic Pt6M (M= Co, Ni, Cu, Zn, Ru, Rh, Pd, Sn, Re, Ir, and Pt). Computational and Theoretical Chemistry. 2023; 1228: 114252.
- Catellani M, Mealli C, Motti E, Paoli P, Perez-Carren˜o E, et al. Palladium-Arene Interactions in Catalytic Intermediates: An Experimental and Theoretical Investigation of the Soft Rearrangement between η1 and η2 Coordination Modes. J. AM. CHEM. SOC. 2002; 124: 4336-4346.
- Marenich AV, Cramer CJ, Truhlar DG. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B. 2009; 113: 6378-6396.
- Tapia O. Solvent effect theories: Quantum and classical formalisms and their applications in chemistry and biochemistry. J. Math. Chem. 1992; 10: 139-181.
- Tomasi J, Persico M, Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994; 94: 2027-2094.
- Tomasi J, Mennucci B, Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005; 105: 2999-3093.
- Reed AE, Weinstock RB, Weinhold F. Natural population analysis. J. Chem. Phys. 1985; 83: 735-746.
- Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital donor-acceptor view point. Chem. Rev. 1988; 88: 899-926.
- Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012; 33: 580-592.