SciBase Journals
SciBase Hematology & Blood Disorders
- Article Type: Review Article
- Volume 1, Issue 1
- Received: Jul 06, 2024
- Accepted: Aug 30, 2024
- Published Online: Sep 06, 2024
Influences on the Hematopoietic Stem Cell Niche
Weiyuan Wang1,2; Timothy J Brown3; Brian M Barth2,4*
1Department of Hematology and Medical Oncology, Winship Cancer Institute, School of Medicine, Emory University, Atlanta, GA 30322, USA.
2Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, Durham, NH 03824, USA.
3Department of Medicine, Division of Hematology and Oncology, University of Texas, Southwestern Medical Center, Dallas, TX 75390, USA.
4Department of Natural Sciences, University of Alaska Southeast, Juneau, AK 99801, USA.
*Corresponding Author: Brian M Barth
Department of Natural Sciences, University of Alaska Southeast, Juneau, AK 99801, USA.
Tel: 907-796-6275;
Email: bbarth@alaska.edu
Abstract
Hematopoietic Stem Cells (HSCs) are supported by the bone marrow microenvironment to maintain normal production of blood cells. The niche may be considered an “ecosystem” that support the function of HSCs and other supportive cells. Alterations in the bone marrow niche are commonly observed in hematologic malignancies. Here, we review recent insights into the location and the molecular and cellular components of the bone marrow niche. Moreover, we discuss how the niche interacts with HSCs to drive the pathogenesis of hematopoietic malignancies. Overall, a better understanding of the influences on the HSC niche may drive therapeutic development targeting defective and aberrant hematopoiesis.
Keywords: Hematopoietic stem cells; Mesenchymal stem cells; Bone marrow; Niche; Microenvironment; CXCL12; Stem cell factor; Thrombopoietin; G-CSF; Angiogenin; Metabolism; Myelodysplastic syndrome; Acute myeloid leukemia.
Citation: Wang W, Brown TJ, Barth BM. Influences on the Hematopoietic Stem Cell Niche. SciBase Hematol & Blood Disord. 2024; 1(1): 1002.
Introduction
Hematopoietic Stem Cells (HSCs) are rare, self-renewing and multipotent progenitors that sustain hematopoiesis throughout the lifespan [1]. This process involves a complex variety of interactions with supporting cells and ultimately generates all types of blood cells, including erythrocytes, platelets, and leukocytes. In this process, HSCs first give rise to an array of hematopoietic stem progenitors which have restricted self-renewing capacity before terminal differentiation into mature cells, such as erythrocytes, megakaryocytes that produce platelets, and leukocytes. In homeostasis, HSCs are mostly in a dormant state, striking a remarkable balance between quiescence and self-renewal. But in cases of hematopoietic stresses such as severe infection or blood loss, HSCs activate to regenerate hematopoietic cells.
HSCs normally reside in the bone marrow within the central cavities of long and axial bones, although they can transiently expand into facultative niches in extramedullary tissues such as the liver and the spleen under severe hematopoietic stresses [1]. The bone marrow microenvironment provides signals for the maintenance and regulation of HSCs, directing proliferation, apoptosis, extra-medullary mobilization, and quiescence [2]. In the past decade, advancements in imaging, HSC biomarker discovery, and improvement in functional genomic techniques have allowed for a deeper understanding of the mechanisms within the bone marrow microenvironment that regulate HSCs. This review will summarize the current understanding of the adult hematopoietic bone marrow microenvironment, focusing on cellular and molecular participants and their contributions to normal HSC regulation as well as the crosstalk between the niche and HSCs in malignant transformation.
The HSC niche
Human bone marrow is located within the trabecular region of the large bones, which is lined by the endosteum, formed from osteoblasts [3,4]. This is highly vascularized and rich in microvessels and capillaries [4,5]. In the center of the bone marrow, longitudinal large arteries give rise to smaller radical arteries, which then branch into arterioles. Arterial circulation transits into venous circulation near the endosteal surface. The network of arterial vessels progressively transforms into wider, irregularly shaped sinusoids which eventually coalesce into a big collecting central sinus and drain into the venous circulation [4,5].
Within the bone marrow, local tissue microenvironments with a distinct cellular and molecular makeup, termed “niches”, maintain and regulate HSCs [6]. The bone marrow is a complex cellular environment with HSCs residing in close contact with multiple cell types, including perivascular mesenchymal stromal cells and endothelial cells [4-7]. These cells secrete growth factors and chemokines that participate in normal hematopoiesis and support progenitor cells. Confocal imaging and spatial modelling studies have confirmed that HSCs are widely distributed throughout the bone marrow but are more common in at the periphery of the marrow near the bone surfaces compared to the medullary regions of the bone, and in the metaphysis of the bone (Figure 1) [8]. In these niches, approximately 80% of HSCs reside adjacent to sinusoidal blood vessels in the bone marrow, the remainder are mainly associated specifically with arterioles or adjacent to transition zone vessels in the endosteal region [1,4,5,9].
Growth factors that modulate HSC function
The normal function of HSCs is supported by cells that reside within the bone marrow niche. These supporting cell types are responsible for the production of growth factors are known to be required for HSC maintenance, including C-X-C motif chemokine 12 (CXCL12), Stem Cell Factor (SCF) and Thrombopoietin (THPO) [1]. Other factors in the bone marrow may also modulate HSC function in a non-cell-autonomous way by promoting hematopoietic regeneration after injury, although they are not necessarily required for HSC maintenance or hematopoiesis.
C-X-C motif chemokine 12 (CXCL12)
CXCL12, also known as stromal cell-derived factor 1, is a chemokine protein ubiquitously expressed in many tissues and cell types and functions as a HSC homing signal to the marrow [3]. In the bone marrow, CXCL12 is constitutively expressed at high levels by perivascular stromal cells. When deleted from mice, absence of CXCL12 is universally fatal shortly after birth [10]. Its receptor, CXCR4, is mainly expressed on immature and mature hematopoietic cell types. This CXCL12/CXCR4 signaling closely regulates HSC retention and maintenance in bone marrow niches [11-13]. Disruption of this connection leads to release of HSCs into the peripheral blood with limited effect on the function of HSC [3,14]. Additionally, depletion of bone marrow macrophages reduces CXCL12 expression in the bone marrow and promotes HSC mobilization and extrameduallary hematopoiesis [15]. Similarly, deletion of Cxcl12 from arteriolar NG2-expressing cells results in HSC reductions and altered HSC localization within the bone marrow [16].
This signaling is also vital for subsequent repopulation of HSCs in the bone marrow [12]. Deletion of Cxcr4 in adult mice resulted in severe reduction of HSC numbers and increased sensitivity to myelotoxic injury [17]. Deletion of Cxcl12 from perivascular stromal cells or endothelial cells depletes HSCs from the bone marrow. In contrast, conditional deletion of Cxcl12from osteoblasts in murine lines leads to loss of lymphoid progenitors but had little or no effect on the number of HSCs [18,19]. Despite this, mice with deletion of Cxcl12 maintain normal levels of B-cell and T-cell progenitors [10]. However, ablation of osteoblasts eventually results in pancytopenia, likely implicating an indirect regulation of HSCs by osteoblasts [20-22]. Indeed, proximal osteolineage cells upregulate genes encoding cell surface and immune response proteins, such as CXCL12 and vascular cell adhesion molecule 1 (VCAM-1), providing a possible mechanism by which osteoblasts can regulate HSCs, however their exact function in hematopoiesis is incompletely understood [18,23].
Stem Cell Factor (SCF)
SCF, also known as KIT ligand, is a growth factor that is required for HSC maintenance. In the bone marrow, SCF is primarily expressed by perivascular and endothelial cells, in proximity to cells that also express CXCL12 [18,20,21]. It activates signaling by the c-KIT receptor tyrosine kinase, which is widely expressed on HSCs and mast cells [24,25]. SCF is present in both membrane-bound and soluble forms [1]. HSCs are depleted in Sl/Sld mutant mice, which lack the membrane-bound SCF but express soluble form [26]. This indicates that the membrane-bound SCF is more important for HSC maintenance. Endothelial and leptin receptor-expressing perivascular stromal cells are the major sources of SCF for HSC maintenance in normal adult bone marrow [20]. Deletion of Scf from each cell population has additive effects on HSC depletion. HSCs are depleted from bone marrow when Scf is deleted from either endothelial cells or leptin receptor-expressing perivascular stromal cells [20]. In this scenario, adipocytes have the capacity to synthesize SCF to maintain the HSC population [27]. In fact, adipocytes in the long bones promote hematopoietic recovery through production of SCF after irradiation [27]. In contrast, HSC frequency or function was no affected by conditional deletion of Scf from osteoblasts, hematopoietic cells, or perivascular mesenchymal stem cells (MSCs) [20].
Thrombopoietin (THPO)
THPO is mostly produced by the liver and kidneys in addition to the bone marrow stroma (to a much lesser extent) is critically involved in the maintenance of adult quiescent HSCs [21,28-30]. Unlike other hormones, THPO is not negatively regulated, rather it is directly regulated by platelet quantities [21,28-30]. In its normal state, THPO produced by the liver promotes megakaryocyte growth, migration and platelet production [31]. Secreted THPO binds to the THPO receptor, which is also known as the myeloproliferative leukemia protein (MPL) and activates a signaling cascade that results in megakaryocyte maturation within the marrow [31]. Maturing megakaryocytes within the marrow interact with numerous cells, including osteoblasts, osteoclasts, HSCs, and plasma cells [31]. Loss of THPO or THPO signaling results in amegakaryocytic thrombocytopenia and eventually progresses to bone marrow failure via loss of HSC quiescence [28,29,32]. Murine THPO-knockout models have demonstrated that absence of THPO results in failure to reconstitute marrow cellularity following irradiation [32]. Further, liver-specific deletion of murine Thpo reduces both marrow megakaryocytes and HSCs without decreasing the overall marrow cellularity, implicating THPO as a critical factor in the maintenance of HSCs [32]. THPO ensures HSC chromosomal integrity and function in response to irradiation by regulating their DNA-damage response [33]. THPO receptor agonists drive HSCs into self-renewing divisions, leading to quantitative expansion of functional HSC [2]. Further, depletion of megakaryocytes results in a loss of HSC quiescence and increases the expansion of HSCs, likely by a concomitant increase in THPO-signaling [34,35]. In ex vivo models, supplementation of HSC cultures with THPO results in accelerated expansion of HSCs [30].
Indeed, during times of hematopoietic distress in which THPO signaling may be enhanced such as during periods of thrombocytopenia or during treatment with THPO-mimetics such as eltrombopag and romiplostim, HSCs exit their quiescent phase and being to divide through an MPL-mediated mechanism [36]. However, long term increases in THPO or treatment with THPO-mimetics results in bone marrow fibrosis through transforming growth factor beta (TGFb), platelet-derived growth factor, and mechanisms resulting in increased bone marrow reticulin production [31,37]. THPO-mediated marrow fibrosis may be reversible upon normalization of THPO levels or cessation of therapy with THPO-mimetics [37].
Angiogenin
Angiogenin is a secreted ribonuclease with a broad range of effects, including angiogenesis, neurogenesis, and immune regulation [28]. In HSCs, angiogenin promotes quiescence, however it promotes proliferation in lineage-committed myeloid progenitors; the exact mechanism is not yet understood [28]. Angiogenin is expressed at a higher level in mesenchymal progenitors, osteolineage-committed progenitors, and periarteriolar sheath cells. It restricts the proliferation of early lymphoid progenitors and HSCs, thereby promoting HSC self-renewal and repopulating potential [29]. Supplemental therapy with angiogenin promotes hematopoietic regeneration following bone marrow failure in stem cell transplantation, while deletion of angiogenin from those cells resulted in an increased number of long-term HSCs and more active cycling of HSCs [28].
Granulocyte Colony Stimulating Factor (G-CSF)
G-CSF is a cytokine that is mainly produced by the endothelium and macrophages. It is clinically used to promote HSC mobilization into the bloodstream and to increase the production of granulocytes, either to reconstitute a depleted immune system or to prepare a patient for a bone marrow donation. To promote mobilization, G-CSF suppresses the expression of CXCL12 from marrow stromal cells, downregulating the homing signal that attracts HSCs to the marrow niche while upregulating CXCR4 [3,10]. Interestingly, activating osteoclasts via administration of RANK-ligand results in mobilization of HSCs, but inhibition of osteoclasts with calcitonin mitigates the G-CSF-induced mobilization of HSCs [3]. It has been hypothesized that activated osteoclasts secrete cathepsin K which may cleave CXCL12 and result in mobilization of HSCs [3]. Further, neutrophils are believed to play a role in G-CSF-mediated HSC mobilization, although this too is incompletely understood [3]. Recently, it was found that these two functions induced by G-CSF work independently. While cells with limited regenerative potential are induced to repopulate by G-CSF, dormant HSCs are only mobilized into the blood without any proliferation [30].
Bone marrow niche cells that regulate HSCs
The HSC niche can be regulated by various types of cells, including perivascular stromal cells and endothelial cells, which can produce factors including CXCL12 and SCF. Several other cell types also contribute to maintenance of the HSC niche [7], and can leverage additional factors including CXCL4, G-CSF, and TGFβ.
Bone marrow stromal cells
CXCL12-abundant reticular cells reside in the marrow at sites closely associated with HSCs [10]. They have been found to largely overlap with nestin-GFP+ stromal cells, and leptin receptor-expressing cells, in studies where both of are defined by transgenic expression using defined stromal-specific promoters [17,20,41-43]. These stromal cells are all MSCs that have both adipogenic and osteogenic potential. They differentiate into adipocytic and osteoblastic mesenchymal lineages in the adult bone marrow, producing large amounts of proteins for the storage of nutritional energy or bone formation [41,43,44]. HSCs are depleted from the bone marrow when Scf or Cxcl12 are conditionally deleted from leptin receptor-expressing CXCL12 abundant reticular cells. This implicates them in a maintenance role for the HSC niche [16,20,44].
Osteoblasts
It had been believed that osteoblasts do not directly promote HSC maintenance as they do not express the crucial niche factors [20,21]. However, evidence that osteoblast ablation eventually causes pancytopenia has led to the hypothesis that osteoblasts indirectly regulate HSCs likely via crosstalk [22]. Recently, stable genome-wide transcriptional differences have been identified by single-cell RNA-seq and transcriptional comparison between HSC-proximal stromal cells and osteolineage cells either in proximity to transplanted HSCs or at a distance. Proximal osteolineage cells displayed a significant upregulation of genes encoding cell-surface proteins and those involved in immune response, such as CXCL12 and VCAM-1, and angiogenin, lending further support for osteoblastic regulation of HSCs [23].
Adipocytes
The bone marrow-resident adipocytes are abundant and their number in the bone marrow increases with age [42]. Moreover, malignant bone marrow stromal cells have been shown to have increased adipogenic potential [45]. Adipocytes have been shown to negatively affect HSC maintenance and are increasingly present after chemotherapy and radiation [46]. Bone marrow adipocytes were found to also synthesize SCF after depletion of endothelial cells and leptin receptor-expressing stromal cells [27]. Despite being an important source of SCF in both locations, adipocytes in long bones promote hematopoietic recovery after irradiation while in caudal vertebrae they inhibit hematopoietic regeneration. Importantly though, adipogenesis can promote initial hematopoietic recovery following irradiation [27]. This is in accordance with the finding that adipocyte-rich bone marrow has decreased numbers of HSCs compared with adipocyte-poor bone marrow [46]. Leukemia cells have been shown to remodel bone marrow adipocytes [47]. This may be critical as different types of adipose tissue, defined as either white, beige or brown, has been shown to be either healthy or associated with inflammation and metabolic disease [48]. Notably, an adipose-regulated pro-inflammatory bone marrow microenvironment has been linked with acquired aplastic anemia [49]. We have also reported a mouse model of obesity and leukemia highlighting the importance of obesity-mediated inflammatory signaling to hematopoietic malignancy development [50].
Endothelial cells
Endothelial cells lining the blood vessels of the bone marrow are also indispensable for HSC maintenance through producing niche factors CXCL12 and SCF and are a central component in the maintenance of these cells [1,19,20,51]. Despite the relatively small amount of CXCL12 and SCF expressed, conditional deletion of Cxcl12 or Scf from endothelial cells has been shown to lead to the decrease of HSC number in bone marrow [19,20]. HSC frequency and function were not affected when Scf was conditionally deleted from other cell types such as osteoblasts, megakaryocytes or other hematopoietic cells [20]. The HSC niche thus depends on SCF or CXCL12 that is produced by endothelial cells and leptin receptor-expressing stromal cells.
Neuronal cells
Bone marrow is highly innervated [13]. Sympathetic nervous fibers generally run alongside arterioles, yet subsets are also found in the sinus wall and the hematopoietic parenchymal tissue, indicating an existence of interconnected network [52]. The nervous system is not required for the maintenance of HSCs in the bone marrow but is critical for bone marrow regeneration after chemotherapy [53]. Signals from the sympathetic nervous system coordinate the circadian egress of HSCs into the circulation by regulating local production of CXCL12 in the synapsed perivascular cells [52,54]. Genetic or pharmacological ablation of adrenergic signaling inhibits G-CSF-induced HSC mobilization [54]. Pharmacological or genetic ablation of adrenergic neurotransmission inhibits G-CSF-induced HSC mobilization [13]. Lastly, nonmyelinating Schwann cells are autonomic nerve-ensheathing glial cells. They are a producer of TGFβ, which is a quiescence signal for HSC [55].
Megakaryocytes
Megakaryocytes are terminally differentiated hematopoietic cells that have an inhibitory role in HSC expansion via the secretion of CXCL4 and TGFβ [34,35]. Studies showed that depletion of megakaryocytes resulted in specific loss of HSC quiescence and led to a marked expansion of functional HSCs. By contrast, conditional deletion of Cxcl4 or Tgfb1 in megakaryocytes increased HSC activation and proliferation [34,35]. While in response to stress, FGF1 signaling from megakaryocytes transiently dominates over TGFβ inhibitory signaling to stimulate HSC expansion to facilitate the recovery of hematopoietic system [34].
Monocytes and macrophages
Higher levels of G-CSF are expressed in monocytes and macrophages as part of the response to sepsis and other inflammatory conditions [56]. Depletion of bone marrow macrophages reduces CXCL12 expression in the bone marrow and promotes HSC mobilization and extramedullary hematopoiesis [15]. Monocytes and macrophages with high expression of αsmooth muscle actin and cyclooxygenase-2 maintain HSCs and protect them from exhaustion during stress situations by producing prostaglandin E2. Moreover, macrophages maintain the quiescence of HSCs through the DARC/CD82 ligand interaction and downstream TGFβ signaling [57].
Sinusoidal and arteriolar niches
Functionally distinct perivascular niches are created by sinusoids and arterioles [8,16,58-60]. There is a higher ratio of quiescent to cycling HSCs associated with arterioles [16,58]. However, overall majorities of both quiescent and cycling HSCs are localized to the sinusoids [8,16,58,59]. The sinusoidal and arteriolar microenvironments differ with respect to vessel wall permeability, oxygen tension and niche factors produced by the residing hematopoietic cells [1,9,16,58,59]. First, sinusoids are very leaky as they have a fenestrated basal lamina. They promote HSC activation and are the exclusive site allowing for cell migration to and from the bone marrow. In contrast, less permeable arterial blood vessels maintain HSCs in a low oxidative state, which can keep HSCs in a quiescent state [59]. They also seem to be more resilient after radiation, thus HSCs may become more dependent on periarteriolar niches during the regeneration of hematopoiesis [61]. Second, direct measurement of local oxygen concentration in the bone marrow showed that the lowest oxygen tension can be found in deeper perisinusoidal regions. The endosteal region, by contrast, is less hypoxic as it is perfused with small arteries [9]. Thirdly, cytokines produced in distinct vascular niches contribute to HSC maintenance differently. Selective Cxcl12 deletion from arteriolar NG2expressing cells caused HSC reductions and altered HSC localization in bone marrow. In comparison, deletion of Scf in sinusoidal leptin receptor-expressing cells led to reductions in bone marrow HSC numbers [16].
Differentiation hierarchy and metabolic features of HSCs
HSCs rely heavily on anaerobic glycolysis and have relatively inactive mitochondria irrespective of oxygen tension, in a manner similar with the Warburg effect in cancer cells [62]. The activity of specific metabolic pathways and the differentiation state of stem cells are clearly related (Figure 2). Cell differentiation is accompanied by a shift from anaerobic glycolysis to mitochondrial respiration; oxygen tension increases from the endosteum to the sinusoids [63,64]. The level of oxygen in the various locations within the bone marrow niche are directly responsible for CXCR4 function and expression, with higher CXCR4 function and expression in relatively hypoxic environments [64]. Moreover, stem cell fate is dependent on the degree of activation of mitochondrial metabolism, in addition to other factors [65]. Initially, embryonic stem cells rely entirely on glycolysis for their source of energy regardless of oxygen availability [66]. After birth, adult stem cells such as HSCs and their progenitor cells reveal a preference for glycolysis and a repression of oxidative phosphorylation [67]. Later, terminally differentiated hematopoietic cells lose their colony-forming capacity and shift from glycolysis to mitochondrial oxidative phosphorylation to produce ATP. In addition, reprogramming somatic cells to a primitive stage results in a metabolic switch from an oxidative to a glycolytic phenotype [68]. These results indicate that the differentiation hierarchy is in close relationship with metabolism, and loss of primitive stem cell potential is accompanied by a remodeling of the mitochondrial compartment. Differentiation of HSCs is accompanied by a biogenic shift from glycolysis to mitochondrial oxidative phosphorylation (Figure 2) [69].
Interestingly, recent studies have demonstrated unique mitochondrial bioenergetic features associated with leukemia and drug resistance [70,71]. Namely, increased oxidative phosphorylation was observed in acute myeloid leukemia (AML) under chemotherapeutic selective pressure but this was not exclusive to the Leukemia Stem Cell (LSC) compartment [70]. This increase in oxidative phosphorylation accompanies increased mitochondrial biogenesis and so has been postulated to be due to an increased reliance on this metabolic pathway [70]. It has therefore been postulated that targeting mitochondrial metabolism may represent a strategy to overcome this aspect of AML drug resistance [70]. However, another study has suggested that increased reliance on oxidative phosphorylation may be representative of mitochondrial inefficiency due to other energetic and metabolic changes [71]. Intriguingly, alterations in mi tochondrial bioenergetics were observed to be associated with and possibly secondary to alterations in sphingolipid metabolism [71]. Not surprisingly, dysfunctional sphingolipid metabolism has also been associated with AML and therapy resistance [71,72]. Overall, this highlights the importance of metabolic changes that occur during hematologic malignancy and demonstrates the relevance of metabolic-targeting therapeutic strategies.
Recent studies by the Jordan group have suggested that LSC metabolism is unique between AML at diagnosis as compared with relapse/refractory AML [73,74]. Specifically, it was shown that amino acid metabolism was upregulated in AML LSCs at diagnosis, but that fatty acid metabolism was favored in relapse/refractory AML LSCs [73]. These results were later expanded on to show that nicotinamide metabolism was upregulated in relapse/refractory AML LSCs [74]. Of note, low levels of Reactive Oxygen Species (ROS) were used as these studies’ primary measure to define LSCs. Therefore, when comparing to other studies it is important to consider that these metabolic findings were uncovered in ROS-low AML LSCs and not AML cellular populations defined in other manners. Notably, the Jordan group’s studies focused on the therapeutic combination of venetoclax with azacitadine [73,74]. Specifically, they showed that this therapeutic combination disrupted various aspects of amino acid metabolism in ROSlow LSCs from AML at diagnosis, including by decreasing the uptake of amino acids [73]. Earlier, the Jordan group had described a role for Bcl-2 as a regulator of amino acid metabolism in ROS-low LSCs and that this represented a metabolic vulnerability in AML [75]. This is intriguing because venetoclax resistance in AML has mostly been attributed to Mcl-1 upregulation as venetoclax is a BH3-mimetic that targets Bcl-2 [76,77]. Previously, the Jordan group showed that Bcl-2 dysregulated oxidative phosphorylation in ROS-low LSCs from AML at diagnosis such as to create a metabolic vulnerability that could be targeted by venetoclax [75]. Interestingly, other recent studies have shown that therapeutics that upregulate or delivery the sphingolipid ceramide can restore AML sensitivity to venetoclax [76,78,79]. Notably, sphingolipids are commonly derived from the amino acid serine, with some exceptions using other amino acids as precursors [80,81]. Therefore, dysfunctional sphingolipid metabolism represents an intriguing and likely important metabolic problem in hematopoietic malignancies.
Malignant transformation of the HSC niche
Hematologic malignancies such as leukemia do not propagate efficiently in vivo outside of the bone marrow and are difficult to grow ex vivo. This suggests that supporting cells within the bone marrow niche may help to support or drive the malignant transformation of HSCs. Furthermore, malignant HSCs may remodel the niche to increase its own support, while suppressing normal hematopoiesis [82-84]. Alteration of the bone marrow niche is an important and necessary step in leukemogenesis [85].
Bone marrow niche contributions to malignant HSCs
Notch signaling in endothelial cells has been shown to lead to the expansion of HSC niches by inhibiting Cxcl12 expression in bone, which increases capillary perivascular cells and arteriole formation and elevates cellular SCF levels [86]. Defects in this signaling pathway have been observed to lead to the development of myeloproliferative disease and has for many decades been linked to the development of T-cell neoplasms [87,88]. FurFurther, it appears that constitutive Notch activation via transduction of HSCs with active Notch1 intracellular domain increases self-renewal of HSCs [88]. Constitutive Notch activation prevents HSCs differentiation into hematopoietic progenitors [88]. To disrupt the cancer-stromal interactions in leukemia, CXCR4 antagonists have been clinically used to promote mobilization leukemic cells from the protective microenvironment, making them more sensitive to conventional chemotherapy [89,90].
Alterations in bone marrow stromal cells have been shown to be sufficient to initiate myeloproliferative disorders [83]. In acute lymphoblastic leukemia and Chronic Myeloid Leukemia (CML), malignant cells were shown to have increased G-CSF production and reduced homing and retention in the bone marrow, which was related to decreased Cxcl12 expression in bone marrow stromal cells [91,92]. Osteogenic differentiation has also been shown to be reduced in myelodysplastic syndrome (MDS)-derived MSC [93]. In fact, MSCs from a wide diversity of MDS subtypes are structurally, epigenetically, and functionally altered, which leads to impaired stromal support and may contribute to deficient hematopoiesis in MDS [93]. An activating mutation of β-catenin in mouse osteoblasts has also been shown to alter the differentiation potential of myeloid and lymphoid progenitors, leading to development of AML. This demonstrates a link between genetic alterations in osteoblasts and the development of AML [94]. Moreover, this shows that an altered microenvironment can serve as an inciting event in hematologic malignancy. Genetic alterations in bone marrow niche cells also indicates that the microenvironment can contribute to the development of myeloproliferative disease, including deletion of Rb, RAR-γ, IκBα, or MIB1 as part of the Notch signaling pathway [95-98]. Additional evidence of the link between changes in the bone marrow microenvironment and malignancy comes from knockdown of Dicer1 in murine MSCs, which was similarly associated with an MDS/AML phenotype [99]. Likewise, knockdown of SBDB in bone marrow niche MSCs was shown to trigger secretion of the damage-associated molecular pattern molecules S100A8 and S100A9 [100]. This is noteworthy because high levels of S100A8 and S100A9 were associated with an increased risk of MDS evolving to AML in patients [100].
In addition, leukemic bone marrow infiltration in an MLL-AF9 AML model has been shown to be promoted by neuropathy of the sympathetic nervous system [101]. Development of AML damages the sympathetic nervous system and disrupts the quiescence of perivascular MSCs. This can lead to increased osteoblastic differentiation at the expense of HSCmaintaining periarteriolar niche cells. Moreover, stromal β2-adrenergic receptors have been shown to regulate LSCs. This is evidenced by rescue of the healthy HSC niche by β2 agonist treatment, which otherwise limits LSC expansion [101].
Malignant HSCs remodel the bone marrow niche
Just as the bone marrow niche may have profound effects on HSC behavior, malignant HSCs may also shape the microenvironment to selectively enhance their own support over normal HSCs [83]. In CML, malignant cells release exosomes that stimulate bone marrow stromal cells to produce IL-8 to promote CML cell survival [102]. The secretion of IL-8 is part of the crosstalk between CML cells and bone marrow stromal cells mediated by exosomes [102]. Furthermore, in myeloproliferative neoplasms, leukemic cells stimulate MSCs to overproduce functionally altered osteoblastic lineage cells, which accumulate in the bone marrow cavity as inflammatory myelofibrotic cells [103]. This is partly due to malignant HSCs that secrete CCL3 and THPO to impact osteoblast function [103]. Malignant HSCs have also been found to produce IL-1β to trigger neural damage and Schwann cell death, which further causes MSC reduction [104]. In addition, MSC derived from MDS or AML patients are altered and do not support normal HSCs indicative of hematopoietic insufficiency within the bone marrow. Further evidence for HSC and MSC crosstalk within the MDS bone marrow microenvironment comes from murine xenotransplantation models. In these models, successful engraftment depended on simultaneous transplantation of both patient-derived MSC as well as patient-derived malignant HSCs [105]. Further indicative of the link between malignancy and the bone marrow niche, in these models MDS patient-derived MSC were also able to reprogram healthy HSCs [105]. Finally, healthy MSCs can take on MDS-like molecular features when exposed to MDS cells, indicative of the ability of malignant cells to directly remodel the microenvironment [105].
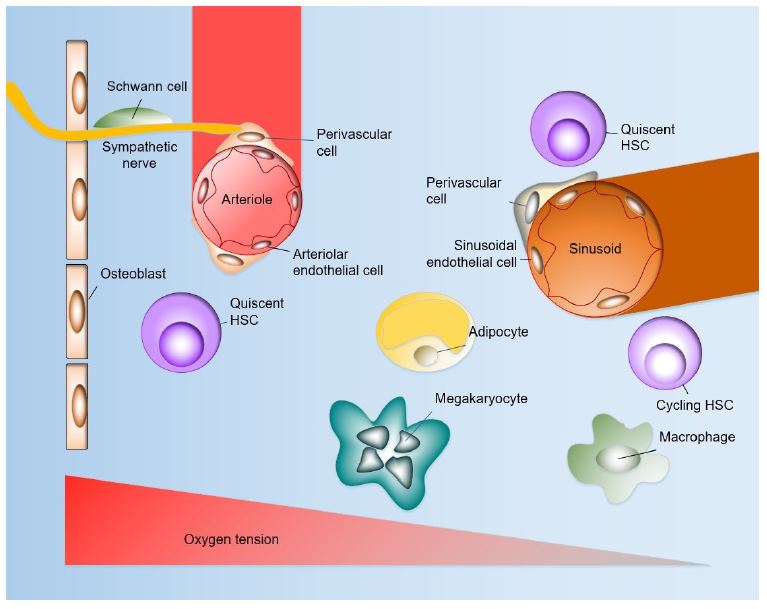
The bone marrow microenvironment is composed of different cell populations that coordinately contribute to the regulation of HSCs. These include perivascular stromal cells, endothelial cells, megakaryocytes, sympathetic neurons and non-myelinating Schwann cells, adipocytes, monocytes and macrophages. The arteriolar niche has a higher ratio of quiescent HSCs, whereas majorities of both quiescent and cycling HSCs localize to the sinusoidal niche. The absolute pO2 of the bone marrow is quite low despite very high vascular density, with the lowest pO2 in perisinusoidal regions. The endosteal region, by contrast, is less hypoxic as it is perfused with small arteries.

Stem cell fate may be directly modified by metabolism. The differentiation of stem cells is accompanied by a biogenic shift from glycolysis to mitochondrial oxidative phosphorylation. HSCs prefer to use glycolysis rather than mitochondrial oxidative phosphorylation as a main energy source. Following differentiation, terminally differentiated cells lose colony-forming capacity and shift from glycolysis to mitochondrial oxidative phosphorylation.
Abbreviations: ESC: Embryonic Stem Cells; iPSC, induced Pluripotent Stem Cells; HSCs: Hematopoietic Stem Cells; HPCs: Hematopoietic Progenitor Cells.
Conclusion
This review has illustrated the contributions of multiple cell populations within the bone marrow microenvironment to the complex regulation of HSC function. There is a continuing and further need to understand how the niche controls HSC function during stress situations, including during infection, malignancy, and during therapy. Examining the crosstalk between HSCs and their niche may provide targetable pathways to alter the HSC niche in such a way that it becomes less hospitable to malignant cells [84]. In this manner, targeting the HSC niche itself is an attractive avenue for the treatment of hematologic malignancies. Future work utilizing high resolution single-cell methods, as well as endogenous labeling and tracking of stem cells, may further foster studies on the complex interplay occurring within the microenvironment of the bone marrow.
Acknowledgements: This work was funded in part by grants from the National Cancer Institute of the National Institutes of Health to BMB (K22-CA190674, R03-CA252825, and P01-CA171983 via subaward from the University of Virginia) and to TJB (T32-CA009679, L30-CA274783), as well as from the National Institute of General Medical Sciences of the National Institutes of Health to BMB (University of New Hampshire COBRE award P20-GM113131, and Alaska INBRE award P20-GM103395). BMB is the owner and founder of Tahosa Bio, LLC (Rapid City, SD). Portions of this work appear in the Ph.D. dissertation of WW (https://scholars.unh.edu/dissertation/2600/) at the University of New Hampshire.
References
- Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. 2017; 17(9): 573-590.
- Geiger H, De Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013; 13(5): 376-89.
- Calvi LM, Link DC. Cellular complexity of the bone marrow hematopoietic stem cell niche. Calcif Tissue Int. 2014; 94(1): 112-24.
- Wang H, Zhang P, Liu L, Zou L. Hierarchical organization and regulation of the hematopoietic stem cell osteoblastic niche. Crit Rev Oncol Hematol. 2013; 85(1): 1-8.
- Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013; 15(5): 533-43.
- Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008; 132(4): 598-611.
- Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011; 208(3): 421-8.
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015; 526(7571): 126-30.
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, Zaher W, Mortensen LJ, Alt C, Turcotte R, Yusuf R, Côté D, Vinogradov SA, Scadden DT, Lin CP. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014; 508(7495): 269-73.
- Hoffman CM, Calvi LM. Minireview: Complexity of hematopoietic stem cell regulation in the bone marrow microenvironment. Mol Endocrinol. 2014; 28(10): 1592-601.
- Walenkamp AME, Lapa C, Herrmann K, Wester HJ. CXCR4 Ligands: The Next Big Hit? J Nucl Med. 2017; 58(Suppl 2): 77S-82S.
- Lai CY, Yamazaki S, Okabe M, Suzuki S, Maeyama Y, et al. Stage-specific roles for CXCR4 signaling in murine hematopoietic stem/progenitor cells in the process of bone marrow repopulation. Stem Cells. 2014; 32(7): 1929-42.
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006; 124(2): 407-21.
- Spiegel A, Kalinkovich A, Shivtiel S, Kollet O, Lapidot T. Stem cell regulation via dynamic interactions of the nervous and immune systems with the microenvironment. Cell Stem Cell. 2008; 3(5): 484-92.
- Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011; 208(2): 261-71.
- Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017; 19(3): 214-223.
- Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006; 25(6): 977-88.
- Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci. 2016; 1370(1): 82-96.
- Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013; 495(7440): 231-5.
- Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012; 481(7382): 457-62.
- Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014; 505(7483): 327-34.
- Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, et al. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004; 103(9): 3258-64.
- Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, et al. Proximity-Based Differential Single-Cell Analysis of the Niche to Identify Stem/Progenitor Cell Regulators. Cell Stem Cell. 2016; 19(4): 530-543.
- Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991; 174(1): 63-71.
- Ho CCM, Chhabra A, Starkl P, Schnorr PJ, Wilmes S, et al. Decoupling the Functional Pleiotropy of Stem Cell Factor by Tuning c-Kit Signaling. Cell. 2017; 168(6): 1041-1052.e18.
- Barker JE. Sl/Sld hematopoietic progenitors are deficient in situ. Exp Hematol. 1994; 22(2): 174-7.
- Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017; 19(8): 891-903.
- Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007; 1(6): 685-97.
- Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007; 1(6): 671-84.
- De Graaf CA, Metcalf D. Thrombopoietin and hematopoietic stem cells. Cell Cycle. 2011; 10(10): 1582-9.
- Khodadi E, Asnafi AA, Shahrabi S, Shahjahani M, Saki N. Bone marrow niche in immune thrombocytopenia: A focus on megakaryopoiesis. Ann Hematol. 2016; 95(11): 1765-76.
- Decker M, Leslie J, Liu Q, Ding L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science. 2018; 360(6384): 106-110.
- De Laval B, Pawlikowska P, Barbieri D, Besnard-Guerin C, Cico A, et al. Thrombopoietin promotes NHEJ DNA repair in hematopoietic stem cells through specific activation of Erk and NF-κB pathways and their target, IEX-1. Blood. 2014; 123(4): 509-19.
- Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014; 20(11): 1321-6.
- D Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014; 20(11): 1315-20.
- Kovtonyuk LV, Manz MG, Takizawa H. Enhanced thrombopoietin but not G-CSF receptor stimulation induces self-renewing hematopoietic stem cell divisions in vivo. Blood. 2016; 127(25): 3175-9.
- Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med. 2009; 60: 193-206.
- Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, et al. Angiogenin Promotes Hematopoietic Regeneration by Dichotomously Regulating Quiescence of Stem and Progenitor Cells. Cell. 2016; 166(4): 894-906.
- Di Scala M, Hidalgo A. Angiogenin Defines Heterogeneity at the Core of the Hematopoietic Niche. Cell Stem Cell. 2016; 19(3): 284-6.
- Bernitz JM, Daniel MG, Fstkchyan YS, Moore K. Granulocyte colony-stimulating factor mobilizes dormant hematopoietic stem cells without proliferation in mice. Blood. 2017; 129(14): 1901-1912.
- Nagasawa T, Omatsu Y, Sugiyama T. Control of hematopoietic stem cells by the bone marrow stromal niche: The role of reticular cells. Trends Immunol. 2011; 32(7): 315-20.
- Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014; 35(1): 32-7.
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010; 33(3): 387-99.
- Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014; 15(2): 154-68.
- Weickert MT, Hecker JS, Buck MC, Schreck C, Rivière J, et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci Rep. 2021; 11(1): 5944.
- Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009; 460(7252): 259-63.
- Yang S, Lu W, Zhao C, Zhai Y, Wei Y, et al. Leukemia cells remodel marrow adipocytes via TRPV4-dependent lipolysis. Haematologica. 2020; 105(11): 25722583.
- Chaurasia B, Talbot CL, Summers SA. Adipocyte Ceramides-The Nexus of Inflammation and Metabolic Disease. Front Immunol. 2020; 11: 576347.
- Gao M, Ge M, Huo J, Ren X, Li X, et al. Leptin-mediated proinflammatory bone marrow environment in acquired aplastic anemia. Cytokine. 2022; 152: 155829.
- Wang W, Sabol RJ, Day AM, Hathorm TG, Clark MN, et al. Obesity Promotes the Ceramide-Mediated NADPH Oxidase in Acute Myeloid Leukemia. J Blood Disorder Mal. 2024; 2(1): 105.
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013; 495(7440): 227-30.
- Giles AJ, Chien CD, Reid CM, Fry TJ, Park DM, et al. The functional interplay between systemic cancer and the hematopoietic stem cell niche. Pharmacol Ther. 2016; 168: 53-60.
- Lucas D, Scheiermann C, Chow A, Kunisaki Y, Bruns I, et al. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat Med. 2013; 19(6): 695-703.
- Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008; 452(7186): 442-7.
- Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011; 147(5): 1146-58.
- Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe. 2014; 15(6): 779-91.
- Hur J, Choi JI, Lee H, Nham P, Kim TW, et al. CD82/KAI1 Maintains the Dormancy of Long-Term Hematopoietic Stem Cells through Interaction with DARC-Expressing Macrophages. Cell Stem Cell. 2016; 18(4): 508-21.
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013; 502(7473): 637-43.
- Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016; 532(7599): 323-8.
- Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016; 530(7589): 223-7.
- Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014; 507(7492): 323-328.
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014; 15(4): 243-56.
- Gaspar JA, Doss MX, Hengstler JG, Cadenas C, Hescheler J, et al. Unique metabolic features of stem cells, cardiomyocytes, and their progenitors. Circ Res. 2014; 114(8): 1346-60.
- Wang A, Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. 2018; 23(10): 729-739.
- Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014; 141(22): 4206-18.
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012; 22(1): 168-77.
- Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013; 32(19): 2589-602.
- Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. 2014; 32(2): 364-76.
- Zhang CC, Sadek HA. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid Redox Signal. 2014; 20(12): 1891-901.
- Farge T, Saland E, de Toni F, Aroua N, Hosseini M, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017; 7(7): 716-735.
- Kao LP, Morad SAF, Davis TS, MacDougall MR, Kassai M, et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL-60 cells. J Lipid Res. 2019; 60(9): 1590-1602.
- Barth BM, Wang W, Toran PT, Fox TE, Annageldiyev C, et al. Sphingolipid metabolism determines the therapeutic efficacy of nanoliposomal ceramide in acute myeloid leukemia. Blood Adv. 2019; 3(17): 2598-2603.
- Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell. 2018; 34(5): 724-740.e4.
- Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell. 2020; 27(5): 748-764.e4.
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013; 12(3): 329-41.
- Barth BM. Ceramide: improving Bcl-2 inhibitor therapy. Blood. 2022; 139(26): 36763678. doi: 10.1182/blood.2022016608.
- Roberts AW, Wei AH, Huang DCS. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood. 2021; 138(13): 1120-1136.
- Lewis AC, Pope VS, Tea MN, Li M, Nwosu GO, et al. Ceramide-induced integrated stress response overcomes Bcl-2 inhibitor resistance in acute myeloid leukemia. Blood. 2022; 139(26): 3737-3751.
- Khokhlatchev AV, Sharma A, Deering TG, Shaw JJP, Costa-Pinheiro P, et al. Ceramide nanoliposomes augment the efficacy of venetoclax and cytarabine in models of acute myeloid leukemia. FASEB J. 2022; 36(10): e22514.
- Ung J, Tan SF, Fox TE, Shaw JJP, Vass LR, et al. Harnessing the power of sphingolipids: Prospects for acute myeloid leukemia. Blood Rev. 2022; 55: 100950.
- Arsenault EJ, McGill CM, Barth BM. Sphingolipids as Regulators of Neuro-Inflammation and NADPH Oxidase 2. Neuromolecular Med. 2021; 23(1): 25-46.
- Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic Stem Cell Niche in Health and Disease. Annu Rev Pathol. 2016; 11: 555-81.
- Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015; 126(22): 2443-51.
- Krause DS, Scadden DT. A hostel for the hostile: The bone marrow niche in hematologic neoplasms. Haematologica. 2015; 100(11): 1376-87.
- Behrmann L, Wellbrock J, Fiedler W. Acute Myeloid Leukemia and the Bone Marrow NicheTake a Closer Look. Front Oncol. 2018; 8: 444.
- Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016; 532(7599): 380-4.
- Lampreia FP, Carmelo JG, Anjos-Afonso F. Notch Signaling in the Regulation of Hematopoietic Stem Cell. Curr Stem Cell Rep. 2017; 3(3): 202-209.
- Weber JM, Calvi LM. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone. 2010; 46(2): 281-5.
- Liu T, Li X, You S, Bhuyan SS, Dong L. Effectiveness of AMD3100 in treatment of leukemia and solid tumors: From original discovery to use in current clinical practice. Exp Hematol Oncol. 2016; 5: 19.
- Tsou LK, Huang YH, Song JS, Ke YY, Huang JK, et al. Harnessing CXCR4 antagonists in stem cell mobilization, HIV infection, ischemic diseases, and oncology. Med Res Rev. 2018; 38(4): 1188-1234.
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012; 21(4): 577-92.
- Van den Berk LC, van der Veer A, Willemse ME, Theeuwes MJ, Luijendijk MW, et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2014; 166(2): 240-9.
- Geyh S, Oz S, Cadeddu RP, Fröbel J, Brückner B, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia. 2013; 27(9): 1841-51.
- Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014; 506(7487): 240-4.
- Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007; 129(6): 1081-95.
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007; 129(6): 1097-110.
- Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha. Immunity. 2005; 22(4): 479-91.
- Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, et al. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 2008; 112(12): 4628-38.
- Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010; 464(7290): 852-7.
- Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell. 2016; 19(5): 613-627.
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014; 15(3): 365-375.
- Corrado C, Raimondo S, Saieva L, Flugy AM, De Leo G, et al. Exosomemediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014; 348(1-2): 71-6.
- Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013; 13(3): 285-99.
- Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014; 512(7512): 78-81.
- Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014; 14(6): 824-37.