
SciBase Journals
SciBase Neurology
ISSN 2691-7785
- Article Type: Case Report
- Volume 1, Issue 1
- Received: Jun 26, 2023
- Accepted: Aug 03, 2023
- Published Online: Aug 07, 2023
Pathomorphological Features of Lhermitte-Duclos Disease: A Rare Clinical Observation and Literature Review
DA Sitovskaya1*; DA Murzaeva1,2,3; Yu M Zabrodskaya1
1Polenov Russian Scientific Research Institute of Neurosurgery-branch of Almazov National Medical Research Centre, 191014, Mayakovskogo st., 12, St. Petersburg, Russia.
2Tyumen State Medical University, Ministry of Health of the Russian Federation, 625023, Odesskaya st., 54, Tyumen, Russia.
3Federal State Budgetary Institution “Federal Center of Neurosurgery” of the Ministry of Health of Russia, 625062, Semovskikh st. 5, Tyumen, Russia.
*Corresponding Author: DA Sitovskaya
Polenov Russian Scientific Research Institute of Neurosurgery-branch of Almazov National Medical Research Centre, 191014, Mayakovskogo st., 12, St. Petersburg, Russia.
Email: daliya_16@mail.ru
Abstract
Dysplastic Cerebellar Gangliocytoma (DCG) or Lhermitte-Duclos Disease (LDD) is a rare neoplasm in the cerebellum of unclear etiology. This pathology can occur as a sporadic disorder or as part of the familial Cowden disease, an autosomal dominant disease characterized by multiple hamartomas involving tissues derived from all three germ cell layers. Given the benign nature and slow growth rate, the prognosis for DCG is predominantly favorable, and surgery is required only if neurological symptoms are present. A clinical case of successful neurosurgical treatment of patient A., 2 years old, in which the disease was manifested by hypertensive-hydrocephalic and convulsive syndrome, is presented. The presented clinical observation of a rare disease acquaints practitioners with the morphological characteristics of the process, which will improve diagnosis and choose the right treatment tactics.
Keywords: Lhermitte-Duclos disease; Granular molecular hypertrophy; Dysplastic cerebellar gangliocytoma.
Citation: Sitovskaya DA, Murzaeva DA, Zabrodskaya YuM. Pathomorphological Features of Lhermitt-Duclauts Disease: A Rare Clinical Observation and Literature Review. SciBase Neurol. 2023; 1(1): 1001.
Actuality
Dysplastic Cerebellar Gangliocytoma (DСM) or Lermitte-Duclos Disease (LDD) is a rare neoplasm of the cerebellum genetically determined by a constitutional mutation of the PTEN (Phosphate and Tensin Homolog) gene. According to the WHO classification of tumors of the central nervous system (CNS) 2021, it belongs to grade 1 (a neoplasm of the CNS with low proliferative activity, slow growth, from cells similar to normal, and rarely spreading to nearby tissues) [1].
J. Lhermitte and P. Duclos first described this tumor in 1920 under the name "diffuse myelinated ganglioneuroma of the cerebellum" in a 36-year-old man, whose initial symptoms appeared at the age of 10 months in the form of hearing loss on the left and pain in the back of the head. The authors came to the conclusion that the pathological formation in the posterior cranial fossa found during autopsy is a combination of a congenital malformation and a tumor frolicking from ganglion cells [2-4]. At the moment, the question remains whether DCM is a tumor or a hamartoma (malformation). Cases of de novo manifestation of the disease, as well as cases of recurrence of education after surgical intervention are described. However, in the overwhelming majority of cases, there is no progression, low proliferative activity, and malformative histological features of the tumor [1].
Lhermitte-Duclos disease can occur as a sporadic disease or as part of the familial Cowden syndrome (OMIM 158350), an autosomal dominant disease characterized by multiple hamartomas involving tissues derived from all three germ layers [5-7]. Approximately 85% of patients with Cowden syndrome have a germline mutation in the PTEN gene (also called "PTEN hamartoma syndrome") located on the long arm of chromosome 10 (10q23.31), including intragenic, promoter mutations, and large deletions/rearrangements [8,9]. The PTEN gene was first discovered as a tumor suppressor in glioma. Researchers associated its somatic mutations with glioblastoma, melanoma, endometrial and prostatic cancer [10]. Patients without PTEN mutations may have SDHB or SDHD germline variants that have been shown to affect the same downstream signaling pathways as PTEN [11].
The leading clinical manifestations of DCM are occlusive hydrocephalus and intracranial hypertension, cranial nerve paresis, and convulsions [1,11-13]. The disease can occur at any age; cases of disease manifestation are known both in early childhood [1,11,13,14] and in elderly patients [1,11,15]. The main method of treatment remains microsurgical removal of the pathological substrate.
Description of the clinical case
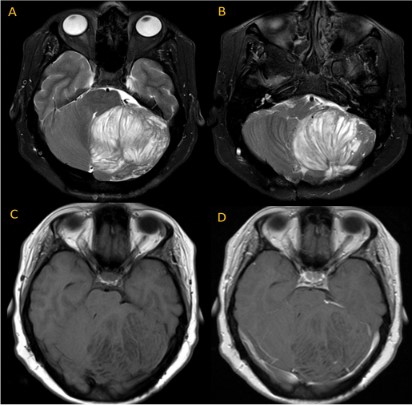
Patient A., female, 2 years old, was admitted to the Federal Center for Neurosurgery in Tyumen. It is known that in the postnatal period, neurological disorders, growth and development disorders were not observed. 4 months before hospitalization, against the background of acute respiratory viral infections, strabismus first appeared, marked weakness, lethargy, convulsive syndrome in the form of twitching of the fingers. A Multispiral Computed Tomography (MSCT) was performed, which revealed a neoplasm of the right cerebellar hemisphere, as well as occlusive hydrocephalus. When performing Magnetic Resonance Imaging (MRI) of the brain with contrast enhancement, a giant tumor of the right cerebellar hemisphere, severe occlusive hydrocephalus with periventricular edema, was verified. Due to the increase in hypertensive-hydrocephalic syndrome, Ventriculo-Peritoneal Shunting (VPS) was performed. After 3 months, the patient underwent a control MRI, which visualized a volumetric formation in the region of the posterior cranial fossa, with a volume of more than 1,5 cm3. No positive dynamics was observed after the VPS. The disease progressed, accompanied by a pronounced dislocation syndrome. The patient was re-admitted for surgery-endoscopic total removal of the cerebellar tumor under neurophysiological control. After the surgical intervention, the patient's condition improved: cerebral symptoms, convulsions and strabismus regressed.
Pathological study
Macroscopic examination of the surgical material showed thickening of the cortex and an increase in the volume of the cerebellar hemisphere.
Histological examination verified the morphological picture of dysplastic cerebellar gangliocytoma, grade 1 (Figure 1, A-J), while the cytoarchitectonics of the cerebellum was preserved (Figure 1A). The neoplasm is represented by large and medium-sized cells with round-oval nuclei and visible nucleoli, with light, abundant eosinophilic cytoplasm, and visible cell processes (Figure 1, B-C). Neuronal tumor cells significantly exceed normal cells in size, Purkinje cells are practically absent, single preserved Purkinje cells are reduced in size. Some of the large cells of the ganglionic type had vitreous opalescent cytoplasm and an eccentrically located nucleus, and some had perimembranous aggregation of Nissl's substance (Figure 1D, G, H). Tumor cells showed a tendency to cluster (Figure 1C). Abundant capillary vasculature was noted (Figure 1E), slight focal lymphocytic infiltration of the tumor stroma, single calcifications. Mitotic activity, necrosis and proliferation of vascular endothelium were not detected. Perifocally, there were fragments of the cerebellum of a typical histological structure. Van Gieson staining showed moderate desmoplasty in the tumor stroma (Figure 1F), Foot’s ammoniacal silver impregnation (Figure 1, I-J) visualized a rich network of reticulin fibers braiding numerous thin-walled capillaries.
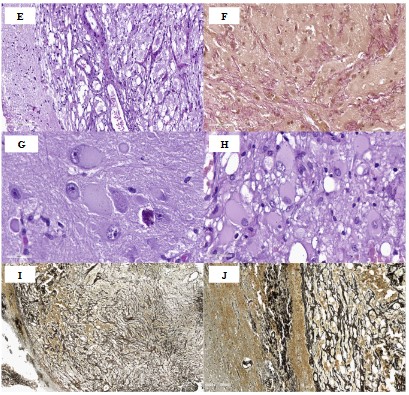
(C,D). Ganglion cells of different sizes, leading to thickening of the gyri of the cerebellum; no Purkinje cells. H&E stain, ×200.
(E). Pronounced capillary vasculature. H&E stain, ×200.
(F). Desmoplasty in the tumor stroma. Van Gieson’s stain, ×200.
(G,H). Ganglion cells with a large round-oval nucleus and a centrally located basophilic nucleolus, abundant opalescent “vitreous” cytoplasm, and perimembranous aggregation of Nissl’s substance. H&E stain, ×400.
(I, J). A rich network of reticulin fibers in the tumor. Foot’s ammoniacal silver impregnation, ×100 and ×200.
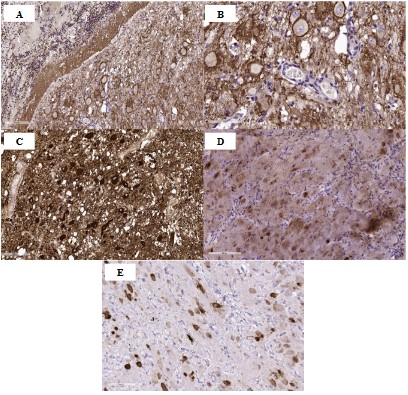
Results and discussion
DCG is characterized by a slow volumetric increase in the cerebellar cortex with the preservation of its laminar histoarchitectonics [1]. Gangliocytoma neurons do not divide, however, the size of the neoplasm can increase slowly due to the growth and myelination of neuronal processes [10]. The main method of diagnosis is radiation research methods: CT and MRI. Thus, the inner part of the lobe of the cerebellum, consisting of white matter, an abnormal granular cell layer, and a ganglionic layer, has a hypointense signal on CT and in T1-weighted MRI, and a hyperintense signal in T2-weighted images [11,18]. The outer part of the petal, consisting of the outer molecular layer and the leptomeninges, is isodense in the projection of the furrows on CT and MRI [16,18]. In addition, in the T2-weighted, a typical "tiger-striped" striation is characteristic – the alternation of internal hyperintense layers and external hypointense layers [12,17,18]. With contrast enhancement, either a weak accumulation of a contrast agent or no accumulation is observed, which, in turn, indicates a slight violation of the blood-brain barrier [12,18].
There is a difference in the genetic type of tumor in children and in adults suffering from this pathology. In adults, the disease is combined with PTEN mutations leading to manifestation in the form of Cowden's disease, and in this case the disease is called COLD syndrome (Cowden-Lhermitte-Duclos syndrome) [19]. There are no mutations in PTEN children. The PTEN gene is a potent tumor suppressor gene (first identified in glioma) [5,12], which encodes a lipid phosphatase for phosphatidylinositol 3-kinase. The latter cleaves phosphates in the inositol ring of proteins and lipids and thus participates in apoptosis [10]. In patients with Lhermitte-Duclos disease, one of the germline alleles of the PTEN gene is likely to be lost, which leads to abnormal growth of cells in the granular layer of the cerebellum [19].
In the given clinical example, the first stage of treatment - ventriculoperitoneal shunting-led to only a slight decrease in the neurological deficit. However, in the course of dynamic observation, a negative trend was noted in the form of a continuing increase in volume formation. A total microsurgical removal of the tumor was performed, which resulted in a complete regression of clinical symptoms.
Conclusion
Lhermitte-Duclos disease is a rare disease, the clinical picture of which is characterized by a slow increase in symptoms, the absence of a clear boundary between the pathological substrate and healthy cerebellar tissue, which is a big problem for surgical resection of the neoplasm and differential diagnosis. This aspect is confirmed by histopathological examination, which reveals the absence of a transition between normal and pathological cortical tissue of the cerebellum. At the moment, there are no uniform approaches to treatment. Given its benign nature and slow growth rate, the prognosis of LDD is predominantly favorable, and surgery is required only if neurologic symptoms are present.
Declarations
Conflict of interest: The author declares no conflict of interest.
Funding: The study was carried out within the framework of State assignment No. 121031000359-3 “Development of new approaches in the diagnosis of mediobasal pharmacoresistant epilepsy based on the histoproteomics of epileptic foci”.
Compliance with patient rights and principles of bioethics: The patient's representatives signed an informed consent to participate in the study.
References
- Louis DN. WHO Classification of tumours editorial board. Central nervous system tumours. Int. Agency Res. Cancer. 2021; 5.
- Lhermitte J, Duclos P. Sur un ganglionneurome diffus du cortex du cervelet. Bull Assoc Fr Etude Cancer. 1920; 9: 99-107.
- Spiegel E. Hyperplasie des kleinhirns. Beitr Pathol Anat. 1920; 67: 539-548.
- Murray C, Shipman P, Khangure M, Chakera T, Robbins P, et al. Lhermitte-Duclos disease associated with Cowden’s syndrome: case report and literature review. Australas Radiol. 2001; 45: 343-346.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012; 18: 400-407.
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997; 16: 64-67.
- Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998; 7: 507-515.
- Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008; 83: 261-268.
- Nieuwenhuis MH, Kets CM, Murphy-Ryan M, Yntema HG, Evans DG, et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer. 2014; 13: 57-63.
- Robinson S, Cohen AR. Cowden disease and Lhermitte-Duclos disease: An update. Case report and review of the literature. Neurosurg Focus. 2006; 20: E6.
- Pritchett PS, King TI. Dysplastic gangliocytoma of the cerebellum-an ultrastructural study. Acta Neuropathol. 1978; 42: 1-5.
- Bimurzin AA, Kalinovsky AV. Surgical treatment of Lhermitte-Duclos disease dysplastic gangliocytoma of the cerebellum. Russian journal of neurosurgery. 2020; 22: 77-82.
- Kumirova E.V. New approaches in the diagnosis of tumors of the central nervous system in children. Russian Journal of Pediatric Hematology and Oncology. 2017; 4: 37-45.
- Roessmann U, Wongmongkolrit T. Dysplastic gangliocytoma of cerebellum in a newborn. Case report. J Neurosurg. 1984; 60: 845-847.
- Gessaga EC. Lhermitte-Duclos disease (diffuse hypertrophy of the cerebellum). Report of two cases. Neurosurg Rev. 1980; 3: 151-158.
- Marcus CD, Galeon M, Peruzzi P, Bazin A, Bernard MH, et al. Lhermitte-Duclos disease associated with syringomyelia. Neuroradiology. 1996; 38: 529-531.
- Saito H, Ogasawara K, Beppu T, Matsuura H, Terasaki K. Biological characteristics of a cerebellar mass regrowing after removal in a patient with lhermitte-duclos disease: emission tomography studies. Case Rep Neurol. 2014; 6: 96-100.
- Lakhani DA, Boo S. Lhermitte-Duclos Disease. Radiology. 2023; 307: e221979.
- Porter S, Cawson R, Scully C, Eveson J. Multiple hamartoma syndrome presenting with oral lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996; 82: 295-301.