
SciBase Journals
SciBase Neurology
ISSN 2691-7785
- Article Type: Case Report
- Volume 1, Issue 1
- Received: Aug 04, 2023
- Accepted: Sep 07, 2023
- Published Online: Sep 12, 2023
Andersen-Tawil Syndrome Associated with Cerebral Cavernoma: Challenge and Diagnostic Delay of a Multisystemic Disease
Susana Arias-Rivas; Manuel Arias*
Neurology Department, University Hospital Complex of Santiago de Compostela, Spain.
*Corresponding Author: Manuel Arias
Neurology Department, University Hospital Complex of Santiago de Compostela., 15706. Santiago de Compostela. Spain.
Email: sakayline@gmail.com
Abstract
Introduction: Andersen-Tawil Syndrome (ATS) is a rare dominantly inherited disorder caused by mutations in the KCNJ2 gene; it presents with cardiac arrhythmias and periodic muscle paralysis, associated with various craniofacial, skeletal and visceral dysmorphias.
Case presentation: A patient, who have been diagnosed with ventricular arrhythmias when she was 5 y o, began to present periodic normokalaemic muscle paralysis when she was 19; as associated malformations, she had clinodactyly of the little fingers and scoliosis. When she was 32 y o, presented a motor seizure, secondary to cerebral cavernoma. The genetic study revealed the p.Thr75Met mutation, in heterozygosis, in the KCNJ2 gene, which was absent in her parents. She has not required treatment for periodic paralysis and the cardiac arrhythmia has been controlled with beta-blockers.
Conclusions: The sequential presentation of ATS manifestations, in a patient with discrete dysmorphias, motivated a delay in the definitive diagnosis; the presentation of epilepsy secondary to cerebral cavernoma had not been referred to date.
Keywords: Muscular periodic paralysis; Cardiac arrhythmia; Andersen-Tawil syndrome; Cerebral cavernoma.
Citation: Arias-Rivas S, Arias M. Andersen-Tawil Syndrome Associated with Cerebral Cavernoma: Challenge and Diagnostic Delay of a Multisystemic Disease. SciBase Neurol. 2023; 1(1): 1003.
Introduction
Andersen-Tawil Syndrome (ATS) is an autosomal dominant inherited genetic disorder (potassium ion channelopathy), described in 1971 by Andersen et al [1]. In 2001, Plaster et al. discovered that it was caused by mutations in the KCNJ2 gene [2]. ATS is one of the hereditary periodic muscle paralysis; this syndrome also includes cardiac arrhythmias (it was named type 7 long QT syndrome) and various craniofacial, skeletal and visceral dysmorphias. The prevalence of ATS is estimated at 0.8-1/1,000,000 and de novo cases constitute 30-50% of the total [3]. The KCNJ2 gene (17q24.3) encodes the synthesis of the alpha subunit of the inward-rectifying potassium channel Kir 2.1, which not only intervenes in the physiology of skeletal and cardiac muscle (it has a key role in the stabilization of the strong negative potential) but it plays an important signaling role during embryogenic development: in the rat embryo, the mRNA of this gene is expressed not only in cardiac and skeletal muscle but also in brain, kidney and tissue craniofacial, vertebral and limb bone [4]. Mutations in other genes, including KCNJ5 that encodes the Kir 3.4 channel, can inhibit the function of Kir 2.1 and cause a syndrome similar to ATS, which is named ATS-2 as there are no mutations in KCNJ2 [5].
The purpose of this paper is the description a case of ATS, a multisystemic disorder that presents with considerable phenotypic variability, even within the same family, and almost half of the cases are caused by de novo mutations: all this explains the difficulties and delays in its diagnosis in the presented patient.
Case presentation
A 38-year-old woman was visited in our department when she was 21 y o. She complained, for about two years, isolated episodes of pain and stiffness, with moderate weakness of the lower limbs, which were triggered after intense exercise, when exposed to cold and in situations of emotional stress; such episodes did not last more than 12 hours and subsided with rest. She was diagnosed, at age 5 years, with cardiac arrhythmia, following admission due to respiratory infection. She had no family history of neuromuscular or cardiological disease.
In the first neurological examination, minimal distal weakness in the lower limbs was observed, with slight difficulty in walking on tiptoe and heels, in addition to hypoactive deep tendon reflexes; Gowers’ sign was negative; her height was 154 cm, and she had slight thoracolumbar scoliosis. In this initial evaluation she underwent: Lab tests (normal ions, minimal elevation of total cholesterol and CK, normal TSH and thyroid hormones); electromyographic study (polyphasic motor potentials in right vastus lateralis and right medial gastrocnemius); 24-hour electrocardiogram-Holter recording (elongated QT, numerous ventricular extra systoles with bidirectional bigeminy and trigeminy); echocardiogram (absence of valvular alteration, normal systolic and diastolic functions; muscular magnetic resonance study (minimal atrophy and fatty infiltration of paraspinal and lower limb musculature); muscle biopsy (small fibers with central nuclei and increased type I fibers, without mitochondrial alterations and with normal histochemical study); negative genetic study of mutations in CACNA1S and SCN4A genes.
In the following years, the patient suffered mild and rare episodes of weakness; cardiological controls were performed without finding any worsening or repercussions of the arrhythmia, for which she received treatment with low doses of beta-blockers (25 mg/day of atenolol). She presented 6 years ago with a left faciobrachial sensory-motor seizure with secondary generalization; a cranial magnetic resonance imaging study detected a right frontal cavernoma (Figure 1A); treatment with levetiracetam was started and she has not had any new seizures. We became aware that she presented discrete clinodactyly of both little fingers (Figure 1B). A new genetic-molecular study detected, in heterozygosis, the p.Thr75Met mutation in the KCNJ2 gene; that mutation was not detected in her parents and in her only sister.
Discussion
The patient described has been finally diagnosed with Andersen-Tawil Syndrome (ATS): Cardiac arrhythmia detected in early childhood, episodes of periodic paralysis starting at the end of the second decade of life, discrete dysmorphologies (short size, scoliosis, clinodactyly of both little fingers) and epilepsy secondary to cerebral cavernoma. She has not presented syncopal symptoms and her cardiac function has remained stable. The association of ATS and childhood-onset epilepsy has been described in 17% of a series of 23 patients [6], but the presence of cerebral cavernomas was not found in a PUBMED search, using both terms.
The causal genetic mutation p.Thr75Met (c.224C>T), is in this case a de novo mutation and has been published in several families: In 2 patients, of a series of 15 cases described by Kostera-Pruszczyk A. et al [7], who had this mutation, the clinical picture of periodic paralysis was not the initial one but both required the implantation of a defibrillator, due to episodes of cardiac arrest.
The neurological symptoms of ATS consist essentially of an episodic picture of periodic paralysis, of greater or lesser intensity, which rarely involves the muscles innervated by the cranial nerves and can present with hyper-, normo- or, more frequently, hypokalaemia. These episodes of muscle weakness and flaccidity may begin as early as infancy, but usually begin in the second decade of life; they are triggered by emotional stress, intense physical exercise, and sometimes prolonged rest and exposure to cold [8,9]. In some patients there is residual muscle weakness that is usually proximal, although in the present case it was distal. It should be noted that syncopal symptoms can be an isolated reason for neurological evaluation and, in these patients, the Electrocardiographic Study (EKG) can be the first clue to reach the diagnosis: the penetrance of cardiological manifestations was 96% in some series [6]. In our case, electrocardiographic alterations were already detected at 5 years of age and the episodes of weakness did not begin until 19 years of age. A few isolated cases of sudden death were reported, a fact that makes it necessary for patients to undergo periodic and exhaustive cardiological controls. Regarding the treatment of acute flaccid paralysis, it will depend on the blood potassium concentration (normal 3.5 to 4.1 mEq/L); if such concentration is low (<3.5 mEq/L), oral potassium administration is usually prescribed (30 mEq/30 minutes without exceeding 300 in 12 hours), with monitoring of EKG and blood levels to avoid hyperkalemia that would worsen the situation. In prophylaxis, lifestyle changes may be sufficient, and depending on the frequency and intensity, acetazolamide or dichlorphenamide are prescribed. It is desirable to maintain blood K levels around 4 mEq/l [8-10)].
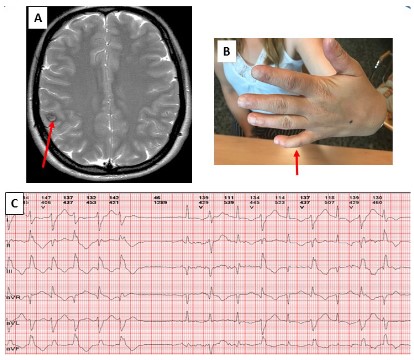
Table 1: Dysmorphias and malformations associated with Andersen-Tawil syndrome: In italics those present in this case.
| Andersen-Tawil syndrome | Malformations/dysmorphias |
|---|---|
| Craniofacial | Scaphocephaly Microcephaly Hypertelorism Wide nose Forehead Low set ears Thin upper lip Prominent frontal sinuses Cleft palate/oval palate Mandibular hypoplasia Maxillary hypoplasia Persistence of primary dentition Decreased number of teeth |
| Skeletal | Short size Small hands and feet Joint laxity Clinodactyly Scoliosis Syndactyly |
| Visceral | Renal hypoplasia Bicuspid aortic valve Coarctation of the aorta Pulmonary artery stenosis Cerebral cavernoma |
Skeletal and visceral dysmorphias vary in number and intensity among different patients and even within the same family (detailed in Table 1). Cognitive impairment, characterized by attention and concentration deficits and executive dysfunction, has been described in some patients [11].
Conclusion
In conclusion, this case shows the sequential presentation of the manifestations of ATS (cardiac arrhythmias and periodic palsy), in a patient with discrete dysmorphias; this fact led to a delay in the definitive diagnosis; although the continued follow-up and cardiological treatment has so far excluded the need for defibrillator implantation; the presentation of epilepsy secondary to cerebral cavernoma had not been referred to date in ATS patients.
Declarations
Declaration of conflicts of interest: None.
Informed consent: Of the patient for publication has been obtained.
References
- Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. 1971; 60: 559-64.
- Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001; 105: 511-9.
- Rajakulendran S, Tam SV, Hanna MG. Muscle weakness, palpitations and small chin: the Andersen-Tawil syndrome. Pract Neurol. 2010; 10: 227-31.
- Adams DS, Uzel SGM, Akagi J, Wlodkowic D, Andreeva V, et al. Bioelectric signaling via potassium channels: a mechanism for craniofacial dysmophogenesis in KCNJ2 associated Andersen-Tawil syndrome. J Physiol. 2016; 594: 2345-70.
- Donaldson MR, Yoon G, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2004; 36: 92-7.
- Haruna Y, Kobori A, Makiyama T, Yoshida H, Akao M, et al. Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum Mutat. 2007; 28: 208.
- Kostera-Pruszczyk A, Potulska-Chromik A, Pruszczyk P, Bieganowska K, Miszcak-knecht M, Bienas P, et al. Andersen-Tawil syndrome: Report of 3 novel mutations and high risk of symptomatic cardiac involvement. Muscle Nerve. 2015; 51: 192-6.
- Nguyen H-L, Pieper GH, Wilders R. Andersen-Tawil syndrome: Clinical and molecular aspects. Int J Cardiol. 2013; 170: 1-16.
- Statland JM, Fontaine B, Hanna MG, et al. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve. 2018; 57: 522-30.
- Pérez-Riera AR, Barbosa-Ramos R, Samesina N, Pastore CA, Scanavacca M, et al. Andersen-Tawil syndrome: a comprehensive review. Cardiol Rev. 2021: 29: 165-77.
- Yoon G, Quitania L, Kramer JH, Fu YH, Miller BL, et al. Andersen-Tawil syndrome. Definition of a neurocognitive phenotype. Neurology. 2006; 66: 1703-10.