
SciBase Journals
SciBase Neurology
ISSN 2996-3788
- Article Type: Case Report
- Volume 1, Issue 2
- Received: Oct 28, 2023
- Accepted: Dec 15, 2023
- Published Online: Dec 22, 2023
Pathomorphology of Metastatic PitNET/Pituitary Carcinoma with Cerebral Dissemination: Autopsy Case Presentation
Darya Sitovskaya1,2,3*; Oleg Verbitskiy3; Yulia Petrova3; Yulia Zabrodskaya1,4
1Polenov Neurosurgical Institute – Branch of Almazov National Medical Research Centre, 197341 St. Petersburg, Russia.
2Department of Pathology with a Course of Forensic Medicine Named after D.D. Lochov, St. Petersburg State Pediatric Medical University, 194100 St. Petersburg, Russia.
3City Mariinskiy Hospital, 191017 St. Petersburg, Russia.
4Department of Pathology, Mechnikov North-West State Medical University, 191015 St. Petersburg, Russia.
*Corresponding Author: Darya Sitovskaya
Polenov Neurosurgical Institute – Branch of Almazov National Medical Research Centre, 197341 St. Petersburg, Russia.
Email: daliya_16@mail.ru
Abstract
Metastatic PitNET(mPitNET) is an extremely rare tumor of neuroendocrine cells of the adenohypophysis with craniospinal and/or systemic metastases and a poor prognosis. The pathogenesis of mPitNET/pituitary carcinoma is not completely clear. Several developmental models have been proposed, including sequential tumorigenesis and de novo transformation. Metastatic PitNETs have been reported in patients with germline tumor predisposition to neoplasia including SDHB mutation and Lynch syndrome. We report a fatal case of a 68-year-old female patient with a third recurrence of null cell PitNET and cerebral dissemination to the pia mater, infiltrative growth into the chiasm, and micro metastasis into the occipital lobe.
Keywords: Metastatic PitNET; Pituitary carcinoma.
Citation: Sitovskaya D, Verbitskiy O, Petrova Y, Zabrodskaya Y. Pathomorphology of Metastatic PitNET/Pituitary Carcinoma with Cerebral Dissemination: Autopsy Case Presentation. SciBase Neurol. 2023; 1(2): 1007.
Introduction
Metastatic PitNET (mPitNET), ICD-O code 8272/3, is a tumor of neuroendocrine cells of the adenohypophysis with craniospinal and/or systemic metastases [1]. mPitNET is a rare type of malignant tumor, rarely diagnosed intravitally and difficult to treat. Most cases mentioned in the literature were diagnosed at autopsy, and delays in diagnosis often have a negative effect on patient outcomes. Even with timely diagnosis, treatment decisions remain difficult in the absence of randomized trials.
Unlike benign “pituitary adenoma,” which is one of the most common intracranial tumors, accounting for 10-15% of intracranial neoplasms [2], mPitNET accounts for only 0.1-0.2% of all pituitary tumors. It can present at any age, but most often appears in the third to fifth decades of life in patients with a pre-existing pituitary adenoma. The prognosis of pituitary carcinoma is generally poor, with overall survival of 2-4 years in patients with CNS metastases and 12 months in patients with systemic metastases [3,4]. Survival after documentation of metastatic disease is poor; 66% of patients die within one year. Treatment options include additional surgery, radiation therapy, and chemotherapy, all of which are associated with poor outcomes. This number has likely improved since the first reported use of temozolamide in 2006; in one meta-analysis including a total of 54 patients, the estimated five-year survival rate was 57.4% for patients with atypical pituitary adenomas and 56.2% for patients with pituitary carcinomas [5]. However, the term “atypical pituitary adenoma” is no longer recommended due to its poor reproducibility and low predictive value.
Histologically, mPitNET/pituitary carcinomas are indistinguishable from PitNET/pituitary adenomas. Histologically, mPitNET/pituitary carcinomas are indistinguishable from PitNET/ pituitary adenomas. Consequently, correctly classifying a lesion as “pituitary carcinoma” can sometimes be a challenging task, only possible in a multidisciplinary team. However, since in the updated 2022 WHO classification of tumors of the endocrine system [1]. All tumors from adenohypophysis cells are assigned /3, this creates predictors for more aggressive primary treatment of all PitNET variants and can serve to increase survival in this group of patients.
Description of the case
Patient V., 68 years old, was admitted to the Mariinsky Hospital in serious condition after being transferred from another hospital. The submitted medical documentation indicated that the patient was being treated for a recurrent pituitary macroadenoma (third relapse) measuring 12×13×18 mm with endo-, supra- and antesellar growth, for which trepanation was performed in the right fronto-parietal-temporal region. The medical history Indicates that Immunohistochemical (IHC) typing of the tumor was performed, based on the results of which nullcell PitNET was verified. It is also known that the woman suffers from non-small cell cancer of the lower lobe of the left lung T4N3M1 with metastatic damage to regional lymph nodes, pleural carcinomatosis and distant metastases to the right lung, for which she underwent video-assisted thoracoscopy on the left with removal of part of the lung lobe and the first line of polychemotherapy. During the examination in the hospital, a multislice computed tomography was performed, the results of which revealed a tumor of the pituitary gland as well as signs of meningoencephalitis. Systemic inflammatory response syndrome was noticeable, with the CRP level being >200 mg/ml, ferritin 711.5 ng/ml, procalcitonin test 12.61 ng/ml, and leukocytosis 41.57x109 /l. The patient was inaccessible to productive contact and their consciousness was at the level of stupor. Complex intensive therapy was carried out in the intensive care unit, however, despite treatment, the woman’s condition remained serious and worsened. On the third day of hospital stay, unfortunately, the patient died.
During the post-mortem autopsy, a sharp exhaustion of the patient was noted, up to cachexia (the thickness of the subcutaneous fatty tissue at the abdominal level was 0.3 cm). Fibrinous pleurisy on the left with obliteration of the pleural cavity was noticeable. A small fragment of a tumor in the lower lobe of the left lung was discovered, and microscopic examination of it revealed moderately differentiated G2 adenocarcinoma of the lower lobe of the left lung with spread to the visceral pleura and metastatic damage to regional lymph nodes and the upper lobe of the right lung; with signs of therapeutic pathomorphosis, ICD-O code 8140/3. Purulent sphenoiditis (Figure 1C), purulent meningo-encephalo-ventriculitis (Figure 1A-B, according to the results of post-mortem bacteriological examination revealed the growth of Klebsiella pneumoniae) were also macroscopically detected and histologically confirmed. In the area of the surgical intervention (area of the sella turcica), a surgical wound with loose edges soaked in blood was found (Figure 1A). Damage of the chiasm was also revealed.
A histological examination of autopsy material from the chiasmal-sellar region revealed a tumor of an alveolar and diffuse structure. The tumor consisted of polymorphic adenocytes with scant to moderate chromophobe cytoplasm and round, hyperchromatic nuclei (Figures 2A-C). Micronecrosis and apoptotic bodies were found in the tumor. The tumor was surrounded by a thin fibrous capsule; foci of growth beyond its boundaries and spread along the soft meninges were identified (Figure 2D). Infiltrative growth into the chiasm (Figure 2E) and micro metastasis in the right occipital lobe (Figure 2F) were also detected.
Thus, the patient with multiple primary malignant tumors in different locations died from edema and displacement of the brain due to purulent meningo-encephalo-ventriculitis, which developed after surgical treatment of a pituitary tumor.
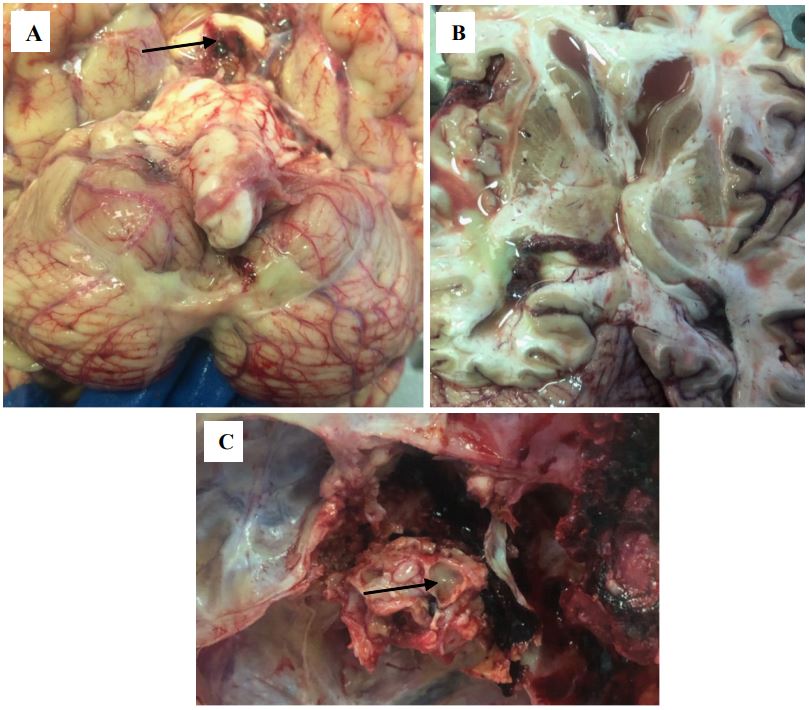
A – Yellow purulent masses in the pia mater of the cerebellum, softening of the chiasm (indicated by the arrow).
B – Purulent masses in the lumen of the lateral ventricles.
C – Purulent masses in the lumen of the sinus of the main bone (indicated by the arrow).
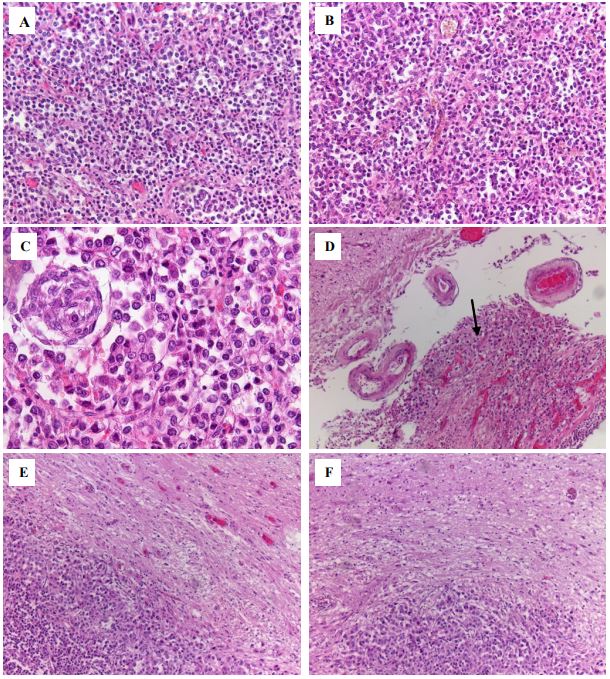
A,B – Tumor of polymorphic adenocytes with alveolar and diffuse type of growth, ×200.
C – Tumor cells form glomeruli, apoptotic bodies are detected, ×400.
D – The tumor spreads through the soft meninges (indicated by the arrow), ×100.
E – Infiltrative growth into the chiasm, ×200.
F – Micro metastasis in the occipital lobe on the right, ×200.
Discussion
The pathogenesis of mPitNET/pituitary carcinoma is not completely clear. Several models have been proposed, including models of sequential tumorigenesis and de novo transformation [6]. In the sequential tumorigenesis model, the development of pituitary carcinoma follows a stepwise transformation of nonadenomatous pituitary cells into adenoma cells, then into an aggressive pituitary adenoma and/or pituitary carcinoma. In the de novo transformation model, pituitary carcinoma cells metastasize from an aggressive pituitary adenoma arising in a normal pituitary gland. Most researchers adhere to the model of sequential oncogenesis with a gradual accumulation of genetic aberrations [7]; however, de novo development cannot be ruled out based on current knowledge.
Currently, there are three possible mechanisms of metastasis in mPitNET/pituitary carcinoma, including droplet metastasis (occurring in the surgical tract), bloodborne spread, and central venous spread [8]. Although tumor cells are shed from the surgical site and drift along the cerebrospinal fluid pathway mainly during surgery, in general mPitNET/pituitary carcinoma tends to metastasize systemically rather than craniospinally. The incidence of systemic metastases is approximately 47%, the incidence of craniospinal metastases is approximately 40%, and the remaining 13% of mPitNETs exhibit both [9]. Different types of mPitNETs have been shown to prefer different patterns of metastatic spread and have their own favorable host organs. Therefore, the “seed and soil” theory may be suitable for the metastatic spread of mPitNETs [10]. Pituitary carcinomas can present clinically in three forms: as incidental findings without symptoms; as functional endocrine neoplasms with secretory symptoms depending on the type of hormone(s) secreted; or as mass lesions with symptoms, including but not limited to headache, vision changes, or double vision [9], often caused by the primary tumor. Most pituitary carcinomas are considered functional, with the most common types being prolactin-secreting and ACTH-secreting carcinomas [11].
Metastases outside the central nervous system are more common than craniospinal metastases in mPitNET. The most common site of distant metastases is the liver, followed by bone, lung, and lymph nodes. The interval between the initial diagnosis of pituitary adenoma and the development of invasive carcinoma is extremely variable, ranging from 4 months to 18 years (mean, 6.6 years; median, 5.0 years) [12]. Metastatic PitNETs have been reported in patients with germline tumor predisposition to neoplasia including SDHB mutation [13] and Lynch syndrome [14], however, there are currently no molecular tests needed to diagnose metastatic pituitary tumors. Aggressive surgical resection of metastases showed improved survival, whereas radiotherapy was only palliative [12]. Future research will focus on identifying those invasive pituitary tumors that are most likely to metastasize and treating them aggressively before they develop into pituitary carcinoma [15].
Institutional review board statement: The work was carried out according to the principles of voluntariness and confidentiality in accordance with Federal Law “On the Basics of Health Protection of Citizens in Russian Federation” 21.11.2011 N 323- FZ, and the Helsinki Declaration on Human Rights.
References
- WHO Classification of Tumours Editorial Board. Endocrine and neuroendocrine tumours. Lyon (France): International Agency for Research on Cancer; 2022. (WHO classification of tumours series, 5th ed.; vol. 10). https://publications.iarc.fr. 2022; 10: 5.
- Shimon I, Melmed S. Management of pituitary tumors. AnnInternMed. 1998; 129: 472-483.
- Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, et al. Pituitary carcinoma: A clinicopathologic study of 15 cases. Cancer. 1997; 79: 804-812.
- Scheithauer BW, Kurtkaya-Yapicier O, Kovacs KT, Young WF, Jr, et al. Pituitary carcinoma: A clinicopathological review. Neurosurgery. 2005; 56: 1066-1074.
- Ji Y, Vogel RI, Lou E. Temozolomide treatment of pituitary carcinomas and atypical adenomas: systematic review of case reports. Neurooncol Pract. 2016; 3: 188-195.
- Di Ieva A, Rotondo F, Syro LV, Cusimano MD, Kovacs K, et al. Aggressive pituitary adenomas--diagnosis and emerging treatments. Nat Rev Endocrinol. 2014; 10: 423-435.
- Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011; 7: 257-266.
- Koyama J, Ikeda K, Shose Y, Kimura M, Obora Y, Kohmura E, et al. Long-term Survival With Non-functioning Pituitary CarcinomaCase Report. Neurol Med Chir. 2007; 47: 475-478.
- Heaney AP. Pituitary Carcinoma: Difficult Diagnosis and Treatment. J Clin Endocrinol Metab. 2011; 96: 3649-3660.
- Dai C, Sun B, Kang J. Is Seed and Soil Theory Suitable for Metastatic Spread of Pituitary Carcinomas? Front Endocrinol (Lausanne). 2021; 11: 607405.
- Scheithauer BW, Fereidooni F, Horvath E, Kovacs K, Robbins P, et al. Pituitary carcinoma: An ultrastructural study of eleven cases. Ultrastruct Pathol. 2001; 25: 227-242.
- Pernicone PJ, Scheithauer BW, Sebo TJ, Kovacs KT, Horvath E, et al. Pituitary carcinoma: A clinicopathologic study of 15 cases. Cancer. 1997; 79: 804-812.
- Tufton N, Roncaroli F, Hadjidemetriou I, et al. Pituitary Carcinoma in a Patient with an SDHB Mutation. EndocrPathol. 2017; 28: 320-325.
- Bengtsson D, Joost P, Aravidis C, et al. Corticotroph Pituitary Carcinoma in a Patient With Lynch Syndrome (LS) and Pituitary Tumors in a Nationwide LS Cohort. J Clin Endocrinol Metab. 2017; 102: 3928-3932.
- Ragel BT, Couldwell WT. Pituitary carcinoma: A review of the literature. Neurosurg Focus. 2004; 16: E7