
SciBase Journals
SciBase Neurology
ISSN 2996-3788
- Article Type: Case Report
- Volume 1, Issue 2
- Received: Nov 07, 2023
- Accepted: Dec 20, 2023
- Published Online: Dec 27, 2023
Atypical Ataxia-Telangiectasia with Cervical Dystonia: A Case Report
Farzad Ashrafi1*; Parisa Azizjalali2; Nasser Moradi3; Bahareh Zaker Harofteh2
1Functional Neurosurgery Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
2Department of Neurological Disease, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
3Department of Neurosciences, School of Science and Advanced Technologies in Medicine, Hamedan University of Medical Sciences, Hamedan, Iran.
*Corresponding Author: Farzad Ashraf
Department of Neurological Disease, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Email: farzad.ashrafi@gmail.com
Abstract
Ataxia telangiectasia is a rare autosomal recessive neurodegenerative disorder. A minority of patients can present with lateonset atypical manifestations due to unknown mechanisms. This can lead to uncommon signs and symptoms, such as dystonic head movements. We report a 26-year-old woman with atypical ataxia telangiectasia who presented with cervical dystonia. Alpha-fetoprotein levels in the serum were found to be elevated in laboratory tests, and genetic testing for the ATM gene provided the definitive diagnosis.
Keywords: Ataxia; Ataxia telangiectasia; Louis-Bar syndrome; Dystonia.
Citation: Ashrafi F, Azizjalali P, Moradi N, Harofteh BZ. Atypical Ataxia-Telangiectasia with Cervical Dystonia: A Case Report. SciBase Neurol. 2023; 1(2): 1008.
Introduction
Ataxia telangiectasia is a rare autosomal recessive neurodegenerative disorder that was initially thought to present exclusively in childhood and primarily characterized by cerebellar symptoms, oculocutaneous telangiectasia, dystonia, parkinsonism, choreoathetosis, myoclonus, tremor, immunodeficiency, cancer susceptibility and radiation sensitivity [1,2]. The incidence of this disease ranges from 1:40,000 to 1: 100,000 [3]. A minority of patients can present with late-onset atypical manifestations due to unknown mechanisms [4]. Here, we describe a 26-year-old Iranian woman with an atypical variant of the disease who presented with late-onset cervical dystonia.
Case presentation
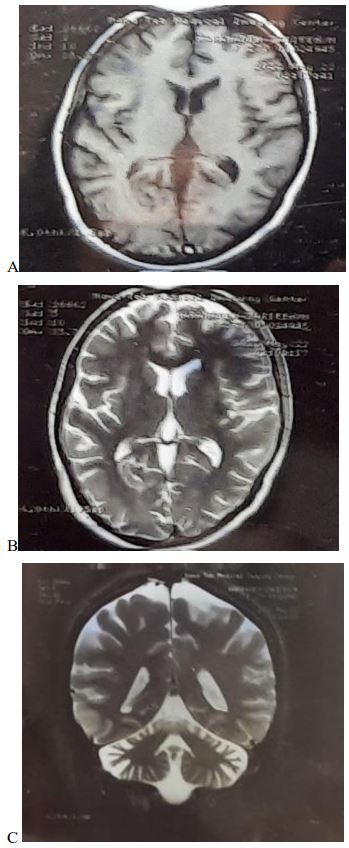
The patient is a 26-year-old woman born as a second child of consanguineous parents. At the age of 20, the patient initially exhibited clinical symptoms in the form of cervical dystonia, which was treated with botulinum toxin injections every 3 months. After two years, she developed a slowly progressive ataxic gaitthat led to repeated falls. There was no history of similar symptoms among her family members. Physical examination demonstrated bilateral horizontal nystagmus, ocular apraxia, distal limbs and cervical dystonia, extrapyramidal symptoms (upper limb’s rigidity, resting tremor, and bradykinesia), and cerebellar ataxia. Cutaneous or ocular telangiectasia was not visible. Montreal cognitive assessment (MOCA) was performed with a score of 26/30. Brain magnetic resonance imaging (MRI) showed cerebellar atrophy (Figure 1). Investigations revealed a high serum Alpha-Fetoprotein (AFP) level. According to the patient’s clinical manifestations, elevated AFP, and presence of a homozygous large deletion spanning exon 59 of the ATM gene, the diagnosis of AT was confirmed. Immunologic and laboratory data are mentioned in detail in Table 1. During follow-up visits, echocardiography reported Mitral valve prolapse and mild Mitral regurgitation without evidence of aorta coarctation.
Table 1: Laboratory data.
| Test | Result | |
|---|---|---|
| WBC | 8.0 1000/yl | |
| Lymphocyte | 33.9% | |
| Neutrophil | 6.2% | |
| Hgb | 15.3 gr/dl | |
| Plt | 252 1000/yl | |
| AST | 18 U/L | |
| ALT | 20 U/L | |
| Calcium | 9.7 mg/dl | |
| Vitamin D | 55.3 ng/ml | |
| Vitamin B12 | 322.50 pg/ml | |
| FBS | 88 | |
| T4 | 9.10 yg/dl | |
| T3 | 1.12 ng/ml | |
| TSH | 2.98 mIU/L | |
| ESR | 4 | |
| CRP | Negative | |
| Iron(Fe) | 71 mic g/dl | |
| Cu (copper) | 112 | |
| Ceruloplasmin | 31 | |
| AFP (alpha-feto protein) | 196.1 ng/ml ( Normal range<6) | |
| Urine | Analysis | Normal |
| 24 hour Cu | 33 | |
| CSF | Glucose | 65 |
| Protein | 479 | |
Discussion
Ataxia telangiectasia (AT), also known as Louis-Bar syndrome, is a rare genetic neurodegenerative disorder. It is the second most common early onset autosomal recessive ataxia after Friedreich’s ataxia. Clinical features are characterized by a combination of neurological and systemic manifestations due to the mutation in ataxia telangiectasia mutated (ATM) gene located on chromosome 11q22-23. This gene encodes a protein kinase with a key role in cell cycle control and DNA repair. Therefore, mutations result in diffuse atrophy of the cerebellum due to loss of Purkinje fibers [3-6]. The prevalence of the disease is between one in 40,000 to one in 100,00 and classically, [5,7]. Symptoms are typically first seen in early childhood, during the time when a child is learning to sit and stand. Classic neurological signs include progressive cerebellar ataxia, oculomotor apraxia, slurred speech, chorea and cognitive dysfunction. Endocrinopathies are common, and extreme sensitivity to ionizing radiation is also part of the clinical picture [1,7,8]. The different presentations in children are categorized as classic/ typical/early-onset in cases where all characteristic features are present, and as variant/atypical/adult-onset in cases presenting with only some of characteristic features, less severe symptoms or late onset of features. In contrast to the classic AT groups, variant AT patients with mild late-onset atypical presentations are characterized by predominance of extrapyramidal features (instead of cerebellar), late onset of ataxia, slower neurologic progression with milder symptoms, and extended lifespan(1,4). Laboratory investigations may reveal elevated levels of serum AFP, low lymphocyte count and other immunological abnormalities. Brain MRI may show cerebellar atrophy. The confirmatory diagnosis is made by cryptogenic and molecular testing of the ATM gene [6,8]. Possible differential diagnoses include ataxia oculomotor apraxia type 1 and type 2, and ataxia-telangiectasia-like disorder (ATLD); However, they can be distinguished from AT by the absence of telangiectasia, normal immunoglobulin and AFP levels, later onset of the condition and slower progression of the disease [9,10]. There is no cure for this condition and genetic counseling should be considered to educate the parents of affected children and identify AT in the antenatal period [6].
We present a rare case of a 26-year-old female who came to the hospital with complaints of dystonic neck movements, which was treated with botulinum toxin injections every 3 months for 2 years and late-onset ataxia which is an uncommon finding at such a young age and she didn’t have any sign of telangiectasia during her illness. This case highlights that diagnosing AT can be challenging due to unpredictable presentations as a result of involvement of multiple systems. Absence of cutaneous and scleral telangiectasia does not oppose the diagnosis of AT, as noticed in our reported case. Patients may exhibit uncommon signs and symptoms, as seen with this case in whom dystonic head motion was the initial presenting sign. Previous reports revealed that within variant atypical AT patients, extrapyramidal movement disorders are the dominant presentation rather than cerebellar signs. Extrapyramidal symptoms commonly predate ataxia in these patients, and the later occurring ataxia is mild to moderate in severity [11]. Dystonia generally appears as a late manifestation and is rarely seen as a presenting feature in classic AT patients. Contradicting a report by Meneret et al. in which dystonia was the presenting feature in 14% of variant patients, only 5% of reported variant atypical AT patients in our literature review presented with dystonia [12,13]. Carrillo et al. reported that patients presenting predominantly with dystonia tend to have a later onset and a milder disease course. Nonetheless, it remains unclear at this time if the dystonic presentation is related to a specific genotypic abnormality [4,14]. Ataxia and telangiectasia may not be prominent features of patients with variant AT treated for dystonia in adulthood, and variant AT may mimic primary torsion dystonia and myoclonus-dystonia [15]. This study supports previous results that AT is not only due to pure ataxia and may appear as only different extrapyramidal signs. As they are likely to be under or misdiagnosed.
Conclusion
Ataxia telangiectasia (AT), also known as Louis-Bar syndrome, is a rare genetic neurodegenerative disorder. Diagnosis of this condition can be challenging as it may present in various forms due to involvement of multiple systems. Some patients may present with uncommon manifestations such as dystonic head movements as seen in our reported case. Absence of cutaneous and scleral telangiectasia does not oppose the diagnosis of AT, as noticed in our reported case. Prognosis is usually poor with increased susceptibility to certain infections and cancers. Proactive measures should be taken for early diagnosis, especially in cases with a family history of AT. Treatment is mostly symptomatic and supportive, and outcomes are generally poor.
Declarations
Author contributions: Farzad Ashrafi: Investigation, Parisa Azizjalali: Visualization, Nasser Moradi: Data curation, Bahareh Zaker Harofteh: Data curation.
Knowledge: None.
Funding information: None.
Conflict of interest statement: The authors declare no conflict of interest.
Data availability statement: The authors confirm that data supporting the funding of this study are available within the article.
Ethics statement: Written informed consent was obtained from sister of the patient to publish this report in accordance with journal’s patient consent policy. Detailed author’s contributed equally to conception, design, manuscript preparation, critical revision, and finalization. All the author agrees to be accountable for all aspects of the work.
References
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: A review. Orphanet J Rare Dis. 2016; 11(1).
- Levy A, Lang AE. Ataxia-telangiectasia: A review of movement disorders, clinical features and genotype correlations - Addendum. Mov Disord. 2018; 33(8): 1372.
- Riboldi GM, Samanta D, Frucht S. Ataxia Telangiectasia. Rosenberg’s Mol Genet Basis Neurol Psychiatr. 2022; 2: 113-6.
- Moeini Shad T, Yazdani R, Amirifar P, Delavari S, Heidarzadeh Arani M, Mahdaviani SA, et al. Atypical Ataxia Presentation in Variant Ataxia Telangiectasia: Iranian Case-Series and Review of the Literature. Front Immunol. 2022; 12.
- Teive HAG, Camargo CHF, Munhoz RP. More than ataxia – Movement disorders in ataxia-telangiectasia. Park Relat Disord. 2018; 46: 3-8.
- Sirajwala AA, Khan S, Rathod VM, Gevariya VC, Jansari JR, Patel YM. Ataxia-Telangiectasia: A Case Report and a Brief Review. Cureus. 2023; 15(5).
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. 2016; 11(1).
- El H, Babikir H. Classic ataxia-telangiectasia in a Sudanese boy: Case report and review of the literature. Sudan J Paediatr. 11(1).
- Bohlega SA, Shinwari JM, Al Sharif LJ, Khalil DS, Alkhairallah TS, Al Tassan NA. Clinical and molecular characterization of ataxia with oculomotor apraxia patients in Saudi Arabia. BMC Med Genet. 2011;12.
- Khan AO, Oystreck DT, Koenig M, Salih MA. Ophthalmic features of ataxia telangiectasia-like disorder. J AAPOS. 2008; 12(2): 186-9.
- Verhagen MMM, Last JI, Hogervorst FBL, Smeets DFCM, Roeleveld N, Verheijen F, et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxiatelangiectasia: a genotype-phenotype study. Hum Mutat. 2012; 33(3): 561-71.
- Mahadevappa M, Santhosh D V, Netravathi M, Yadav R, Pal PK, Ravi Y, et al. Clinical profile of hundred patients with ataxia telangiectasia from India. Park Relat Disord. 2016; 22: 153-4.
- Méneret A, Ahmar-Beaugendre Y, Rieunier G, Mahlaoui N, Gaymard B, Apartis E, et al. The pleiotropic movement disorders phenotype of adult ataxia-telangiectasia. Neurology. 2014; 83(12): 1087-95.
- Carrillo F, Schneider SA, Taylor AMR, Srinivasan V, Kapoor R, Bhatia KP. Prominent oromandibular dystonia and pharyngeal telangiectasia in atypical ataxia telangiectasia. Cerebellum. 2009; 8(1): 22-7.
- Saunders-Pullman R, Raymond D, Stoessl AJ, Hobson D, Nakamura T, Pullman S, et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian Mennonites. Neurology. 2012; 78(9): 649-57.