
SciBase Journals
SciBase Neurology
ISSN 2996-3788
- Article Type: Case Report
- Volume 2, Issue 1
- Received: Mar 20, 2024
- Accepted: Apr 23, 2024
- Published Online: Apr 30, 2024
Massive Cerebral Edema in Hypertensive Emergency with Complete Neurological Recovery: Atypical PRES? A Case Report
Eric RA Laldjising*; Jhuresy E La Roche; Joris AJ Osinga; Iris JAM Verberk Jonkers; Walid Moudrous
Department of Neurosurgery, Park Medical Center, Rotterdam, The Netherlands.
*Corresponding Author: Eric RA Laldjising
Department of Neurosurgery, Park Medical Center, Rotterdam, The Netherlands.
Email: ericlaldjising@hotmail.com
Abstract
Objective: We report a case of a 38-year-old female with massive cerebral edema caused by hypertensive emergency with full neurological recovery, fitting the criteria of posterior reversible encephalopathy syndrome. Genomic sequencing revealed a possible pathogenic complement factor mutation.
Data sources: Maasstad Hospital Rotterdam
Study selection: Case report.
Data extraction: Clinical records.
Data synthesis: None.
Conclusion: Our case with massive cerebral edema with compromised ventricles highlights the importance of a broader clinical and radiological definition of posterior reversible encephalopathy syndrome. Furthermore, we question the indication for monitoring and treatment of elevated intracranial pressure in hypertensive emergency.
Keywords: Hypertensive encephalopathy; Posterior reversible encephalopathy syndrome; Intracranial pressure; Atypical hemolytic uremic syndrome.
Citation: Laldjising ERA, La Roche JE, Osinga JAJ, Verberk Jonkers IJAM, Moudrous W. Massive Cerebral Edema in Hypertensive Emergency with Complete Neurological Recovery: Atypical PRES? A Case Report. SciBase Neurol. 2024; 2(1): 1013.
Case presentation
A 38-year-old Caucasian female presented at the emergency department with progressive apathy, decreased muscle strength resulting in difficulty walking, headache, chest pain and blurry vision. The patient reported neglected hypertension for several years and was non-compliant with previously prescribed metoprolol. No substance abuse was reported. Maternal family history was significant for hypertension with no known secondary cause. Symptoms developed gradually in prior weeks and were rapidly progressive 24 hours before presentation.Physical examination showed an initial blood pressure of 268/164 mmHg and a pulse of 65 beats per minute. Patient was lethargic with a decreased EMV score (E3M5V4) and no lateralization or focal neurological signs. Further physical examination was unremarkable, with no heart murmurs or signs of (pulmonary) edema.
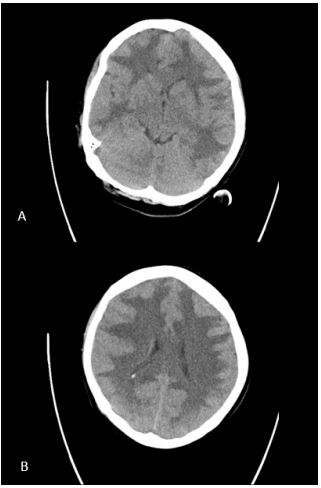
Electrocardiogram displayed all criteria’s for left ventricular hypertrophy which was later confirmed by additional echocardiography. Laboratory findings at admission were concerning, manifesting renal failure (creatinine 884 µmol/L, urea 39.4 mmol/L) and hemolytic anemia (hemoglobin 5.7 mmol/L, thrombocytes 146x10^9/L, haptoglobin < 0.1 g/L, LDH 723 U/L). The urine analyses showed 4+ hemoglobin, 3+ protein, protein/ creatinine ratio of 294 mg/mmol. Head CT-scan revealed massive cerebral edema with compromised ventricles, loss of grey white matter differentiation more distinct in the right parietal and frontal lobe (Figure 1). Further workup showed grade IV retinopathy with papillary edema.
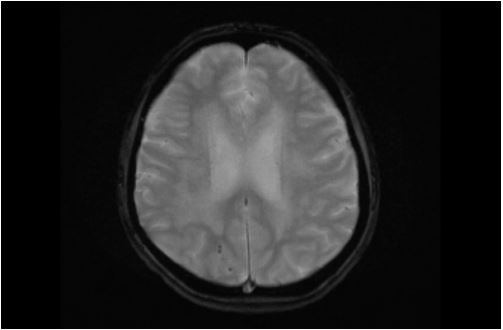
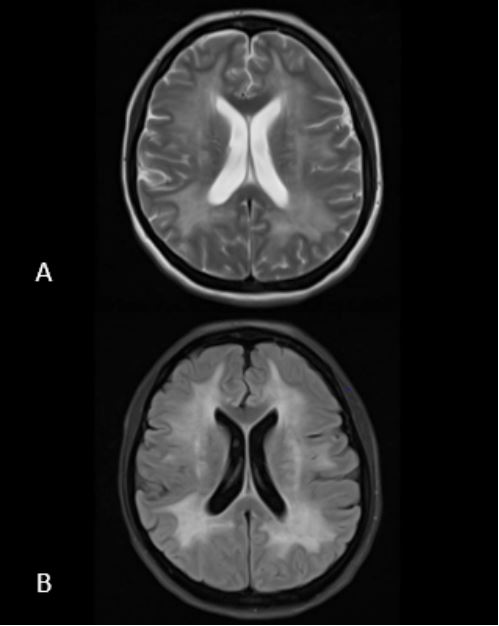
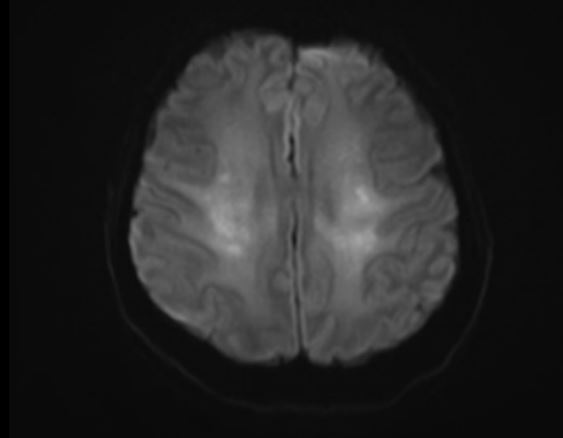
The patient was urgently admitted to the intensive care unit for blood pressure lowering treatment and monitoring. Taking into account her neurological symptoms and lethargic state with the impressive occurrence of cerebral edema, all consistent with severe hypertensive encephalopathy, intracranial pressure monitoring was considered but not applied. The patient was put in a constant Semi-Fowler’s position. Treatment initially consisted of intravenous labetalol and oral calcium channel blocker, aiming for a systolic blood pressure reduction of 25% in the first 24 hours. Due to progressive oliguria along with inadequate decrease in mean arterial pressure, intravenous diuretics were added to the treatment regimen. Despite all efforts the renal function declined requiring continuous veno-venous hemofiltration days after admission, with optimal blood pressure treatment consisting of a beta blocker, calcium channel blocker, ACE inhibitor, lisdiuretic and potassium sparing diuretic. A second head CT-scan three days later showed normal ventricle sizes suggesting decrease in cerebral edema. Grey white matter differentiation remained unaffected. About a week after admission periventricular leukoencephalopathy was still evident on MRI with small areas of diffusion restriction and micro hemorrhages in the right parieto-occipital lobe and left temporal lobe (Figure 2A-C). Neurological symptoms improved over the first week (headache, blurry vision) and eventually fully disappeared after several weeks. MRI findings have either significantly improved or normalized with no signs of cerebral edema and periventricular white matter lesions which in part improved in comparison with previous MRI (Figure 3A-C).
In conclusion, we present a case of massive hypertensive emergency with encephalopathy and possible signs of Posterior Reversible Encephalopathy Syndrome (PRES), Thrombotic Microangiopathy (TMA) with renal failure and early signs of heart failure. All symptoms reversed after treatment with antihypertensive agents, except renal failure.
Background
Hypertensive emergency is a condition defined by severely elevated blood pressure, typically systolic pressure above 200- 220 mmHg and/or a diastolic pressure above 120 mmHg [1]. Malignant hypertension is a general term that is used to describe this condition.
Hypertensive emergency is characterized by symptoms corresponding with either hypertensive encephalopathy, (sub) acute kidney injury, signs of heart failure or acute myocardial ischemia, aortic dissection, thrombotic microangiopathy or retinopathy. Blood pressure reduction should be gradual, with a maximum decrease in mean arterial pressure of 25% in the first 24 hours, ideally with 10% to 20% in the first hour [2]. Accelerated decline in blood pressure, exceeding a decrease >50% of the mean arterial pressure, can result in cerebral ischemia and even death.
Hypertensive Encephalopathy (HE) is a rare condition and is a diagnosis per exclusionem after other causes of neurological deficits are ruled out in the presence of hypertension. It is caused by sudden or severe increase in blood pressure and as a result failure in autoregulation. With increased blood pressure, arteriolar vasoconstriction occurs resulting in a rise in vascular resistance causing damage to the vascular wall and release of cytokines, ultimately leading to a breakdown in the blood-brain barrier which induces cerebral edema and petechial micro hemorrhages [3]. Hypertensive encephalopathy occurs in approximately 10-15% of the patients with malignant hypertension.
PRES is a combination of symptoms such as altered mental status, headache, visual changes and seizures, with typical imaging findings of cerebral edema in the postero-occipital lobe. Unlike the name suggests, other areas can also be affected as reported in more recent studies [4,5].
HE can either be present in the absence of typical radiographic findings or as a manifestation of PRES. In both illnesses, extensive cerebral edema is indicative of increased intracranial pressure that, if left untreated, can lead to secondary brain injury or herniation. Monitoring and therapeutic options for elevated intracranial pressure have been widely researched, but mostly in patients with traumatic brain injury [6]. It is unclear if intracranial pressure monitoring for HE is beneficial or if additional treatments may lead to a better neurological outcome.
Differential diagnosis
Thorough workup was done for exclusion of secondary causes of hypertensive emergency. Hydronephrosis, renal artery stenosis and renal fibromuscular dysplasia were excluded with CT angiography. Endocrinological tests were negative. Respective serum antibodies were negative for renal antiphospholipid syndrome and there were insufficient criteria for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura.
Atypical Hemolytic Uremic Syndrome (aHUS) is an uncommon progressive condition that manifests because of complement system activation, typically by the alternative pathway [6]. This is triggered by genetic predisposition or other complementamplifying conditions such as, autoimmune diseases, pregnancy complications or malignant hypertension. The latter is also a known complication of aHUS, making it difficult to determine the primary diagnosis. aHUS is characterized by endothelial damage due to increased activation of complement factors, antibodies against factor H or increased intrinsic activity of complement C3 or factor B [7]. Serum level for complement factor C3 in this patient was slightly below threshold 0.82 g/L, (0.84-1.68g/L), but is not considered a reliable diagnostic. Complement activation markers were determined and Bb and C3bBbP were significantly elevated, 2.83 µg/ml (0-1.84 µg/ml) and 74.6 CAU/ml (0-12 CAU/ml). C3bc and C5b-9 (TCC) were mild to moderately elevated, 15.7 CAU/ml (0-15 CAU/ml) and 0.79 CAU/ml (0-0.5 CAU/ ml). Other complement activation markers (C1 inhibitor-C1 rs complex, C5a, C3bc and C4DP) were all within normal range. Antibodies against complement factor H were not detected. Genomic sequencing for known pathogenic mutations revealed a heterozygosity sequency variant c.2363C>T p. (Thr788Met) in this patient. According to prediction softwares (Align GVGD, SIFT and PolyPhen) it could be pathogenic for C3 genome and has a population prevalence of 0,0021%. Hypertensive emergency is a common cause of TMA and genetic complement mutations are often associated in these cases [7]. There have also been reports of PRES manifestations in aHUS [8]. Considering all results in this case of severe hypertensive emergency with TMA and possible PRES, these findings support a possible pathogenic mutation of complement factor C3 as underlying cause.
Discussion
Unlike the name suggests, other areas can also be affected in PRES as reported in recent studies. Frontal and temporal lobe in up to 75%, cerebellum in 50% of the cases and basal ganglia and brainstem are other areas of involvement in one third of the cases [4,5]. There are several known conditions that can cause PRES, HE being one of them [5]. Our case of massive cerebral edema and micro hemorrhages with almost complete reversal on MRI scan, in our opinion, can only be attributed to an atypical manifestation of PRES in hypertensive encephalopathy. PRES comprises a broad spectrum of clinical presentation and radiological characteristics [4]. Renaming this entity and adjusting the specific characteristics might be appropriate for proper clinical diagnosis and treatment.
Furthermore, despite complete resolution of neurological symptoms after antihypertensive agents were administered, we cannot help but wonder if in relation to the manifest massive cerebral edema Intracranial Pressure (ICP) monitoring and lowering would have been of added value in this case. To our knowledge, there are no studies that report the significance of ICP monitoring or additional treatment for elevated intracranial pressure in HE or as a manifestation of PRES. In Griswold et al. [9], a case series of 3 children with HE in comatose state is described where ICP monitoring and cerebral perfusion pressure targeted therapy were applied. Other means of decreasing intracranial hypertension were also used. However, from this case series alone no conclusions can be drawn regarding significance of ICP monitoring. It is also unclear if there is a real time correlation between ICP and mean arterial pressure. For Traumatic Brain Injury (TBI), ICP monitoring has been extensively researched and guidelines recommend it with specific targets for blood pressure and cerebral perfusion pressure [6]. However, it is not known if the same can be applied for hypertensive encephalopathy, especially since the pathophysiology is entirely different [6]. Hypothetically in the case of HE, ICP is highly associated with the mean arterial pressure and ICP monitoring would be less beneficial then in TBI. Furthermore, the gold standard for monitoring ICP is an intraventricular catheter, which is an invasive procedure with risk of adverse events.
Also, there are several ways to manage increased ICP. Optimal blood pressure, glycemic control and keeping a core temperature of maximum 37 degrees are simple noninvasive methods with clear benefit [6]. Other possible interventions are head elevation up to 30 degrees and induced hyperventilation. Both can cause a rebound effect if executed incorrectly, respectively elevation above 30 degrees and hyperventilation longer than 24 hours [6]. The use of mannitol and hypertonic saline have been researched in traumatic brain injury showing no difference in neurological outcome between the two [6]. In our case, both therapies are relatively contra-indicated due to the existence of renal failure.
The association between genetic complement mutation and hypertensive emergency with TMA is not unusual. There are numerous factors supporting aHUS as underlying cause. The patient’s family history, the combination of severe hypertensive emergency with TMA, renal failure persisting after optimal blood pressure, the association with PRES and the possible pathogenic C3 mutation. There was a delay in diagnosis as a result of transfer from a smaller hospital to our dialysis centre a few days after admission. Workup including complement markers was not completed until roughly two weeks later. Around that time the patient was relatively stable on dialysis and was discharged against medical advisement pending genome sequencing. Therefore, she was not treated with neither plasmapheresis or eculizumab for aHUS.
Conclusion and follow up
There is a broad spectrum of clinical and radiological characteristics that correspond with posterior reversible encephalopathy syndrome. The current definition may leave certain patients undiagnosed and possibly untreated, therefore renaming and adjustment of the diagnostic criteria may be appropriate. Nowadays, there is no evidence supporting routine ICP monitoring and treatment in hypertensive encephalopathy. However, if there are no signs of improvement with antihypertensive treatment alone, ICP monitoring and treatment might be considered.
Currently, our case-patient is dialysis-dependent and is in the process for renal transplant screening taking into account possible recurrence of aHUS after transplantation. At the moment there are no neurological deficits. Fundus photography is still slightly abnormal with remaining macular exudates.
References
- Van den Born BH, Lip GYH, Brguljan-Hitij J, et al. ESC Council on hypertension position document on the management of hypertensive emergencies [published correction appears in Eur Heart J Cardiovasc Pharmacother. 2019 Jan 1;5(1):46]. Eur Heart J Cardiovasc Pharmacother. 2019;5(1):37-46. doi:10.1093/ehjcvp/pvy032
- Elliot WJ: Clinical features in the management of selected hypertensive emergencies. Prog Cardiovasc Dis. 2006; 48: 316-325.
- Miller JB, Suchdev K, Jayaprakash N et al: New developments in hypertensive encephalopathy. Curr Hypertens Rep. 2018; 20: 13.
- Fugate JE, Rabinstein AA: Posterior reversible encephalopathy syndrome: Clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet neurol 2015; 14: 914-25.
- Tetsuka S, Ogawa T. Posterior reversible encephalopathy syndrome: A review with emphasis on neuroimaging characteristics. J Neurol Sci. 2019; 404: 72-79.
- Schizodimos T, Soulountsi V, Iasonidau C. An overview of management of intracranial hypertension in the intensive care unit. J Anesth. 2020; 34: 741-747.
- Cavero T, Arjona E, Soto K, et al. Severe and malignant hypertension are common in primary atypical hemolytic uremic syndrome. Kidney Int. 2019; 96: 995-1004.
- Medeni SS, Namdaroglu S, Cetintepe T, et al. An adult case of atypical hemolytic uremic syndrome presented with posterior reversible encephalopathy syndrome: Succesful response to late-onset eculizumab treatment. Hematol Rep. 2018; 10: 7553.
- Griswold WR, Viney J, Mendoza SA, et al. Intracranial pressure monitoring in severe hypertensive encephalopathy. Crit Care Med. 1981; 9: 573-576.