
SciBase Journals
SciBase Neurology
ISSN 2691-7785
- Article Type: Research Article
- Volume 2, Issue 2
- Received: Jun 06, 2024
- Accepted: Jul 15, 2024
- Published Online: Jul 22, 2024
Modifications on Histone Tails in Parkinson’s Disease
Qiao Mao1#; Zhixiong Luo2#; Kesheng Wang3 ; Bin Chen4 ; Zhiren Wang5 ; Yong Zhang6 ; Xiaoping Wang7*; Xingguang Luo5,8*
1Department of Psychosomatic Medicine, People’s Hospital of Deyang City, Deyang, Sichuan 618000, China.
2College of Integrative Medicine, Fujian University of Traditional Medicine, Fuzhou 350122, China.
3Department of Family and Community Health, School of Nursing, Health Sciences Center, West Virginia University, Morgantown, WV 26506, USA.
4Department of Cardiovascular Medicine, Shengli Clinical Medical College of Fujian Medical University, Fujian Medical University, Fujian Provincial Hospital, Fuzhou, Fujian 350001, China.
5Beijing Huilongguan Hospital, Peking University Huilongguan Clinical School of Medicine, Beijing 100096, China.
6Institute of Mental Health, Tianjin Anding Hospital, Mental Health Center of Tianjin Medical University, Tianjin 300222, China.
7Department of Neurology, Jiading Branch of Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
8Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA.
#These authors contributed equally.
*Corresponding Author: Xiaoping Wang
1Department of Neurology, Jiading Branch of Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
Email: x_p_wang@sjtu.edu.cn
Xingguang Luo
2Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA.
Email: Xingguang.Luo@yale.edu
Abstract
This study investigates the role of histone tail modifications in Parkinson’s Disease (PD), emphasizing the epigenetic regulation of genes associated with the disease. PD primarily manifests in individuals over 60, suggesting that PD-causal genes remain dormant until later in life, influenced by environmental factors and epigenetic modifications. Histone modifications such as methylation, acetylation, phosphorylation, and ubiquitylation play crucial roles in gene expression regulation by altering chromatin structure or interacting with gene regulatory regions. Specifically, modifications on histones H2A, H2AX, H3, and H4 have been linked to PD. For instance, α-Synuclein (α-SYN) aggregation, a hallmark of PD, is regulated by histone modifications like H3K27ac and H3K4me3, which enhance α-SYN expression and contribute to PD progression. Conversely, repressive marks like H3K9ac and H3K27me3 can mitigate PD risk by reducing α-SYN levels. Therapeutic strategies targeting these histone modifications, such as the use of GSK-J4 or vitamin C-treated neural stem cells, show potential in alleviating PD symptoms by modulating histone marks and gene expression. Understanding these epigenetic mechanisms offers promising avenues for developing novel treatments for PD.
Keywords: Histone tail; Histone modification; Parkinson’s disease; Methylation; Acetylation; Epigenetic regulation; Gene expression.
Citation: Mao Q, Luo Z, Wang K, Wang X, Luo X, et al. Modifications on Histone Tails in Parkinson’s Disease. SciBase Neurol. 2024; 2(2): 1017.
Introduction
Parkinson’s Disease (PD) predominantly manifests after the age of 60, with less than 10% of cases occurring before 50 [1]. This raises the question: if PD is caused by genes present from birth whose sequences remain unchanged throughout life, why doesn’t it develop during infancy, childhood, or adolescence? The straightforward answer is that PD-causal genes do not express before the age of 60. Thus, alongside genetic research that identifies PD-causal genes, epigenetic research exploring the regulation of these genes is crucial.
Human DNA, though 6 feet long, is not linear but densely folded. It wraps around histones to form nucleosomes, which are then packed into 30-nanometer fibers, forming chromatin, and further condensed into chromosomes. This compact folding may protect genes from transcription until influenced by environmental factors, including gene expression regulators like histone tail modifications.
Histones act as spools around which the DNA double-helix wraps twice to form nucleosomes. The nucleosome core is an octameric structure composed of two H2A-H2B dimers and one H3-H4 tetramer. Each histone has N-terminal and C-terminal tails that can unwind or condense chromatin [2] and strengthen or disrupt histone-DNA interactions, thereby enhancing or repressing gene expression. Compared to the nucleosome core, the more flexible tails are especially susceptible to modifications by medicines, drugs, compounds, or other environmental factors.
The length of these tails is limited. For instance, the N-terminal tails of H2A, H2B, H3, and H4 consist of 23, 35, 40, and 25 amino acids, respectively. PD-related modifications include methylation, acetylation, phosphorylation, and ubiquitylation, with lysine (K) being the most commonly modified amino acid. In total, there are 15 histone tail modifications associated with PD: one on H2A, two on H2AX, ten on H3, one on H4, and none on H2B (Figure 1). This study summarizes the role of histone tail modifications in regulating PD-associated genes.
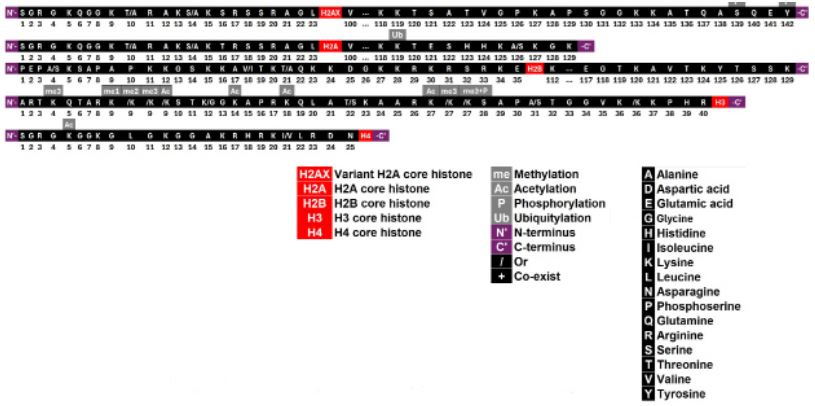
Methodology
A systematic literature search was conducted using the PubMed database to identify papers published up to June 2024, with the search terms “H2A* OR H2B* OR H3* OR H4*” and “Parkinson’s disease.” The study examined over 200 common histone modifications. The present study reviewed, extracted, summarized, illustrated, and discussed the positive findings linking histone modifications to Parkinson’s disease.
Results and discussions
α-synuclein (α-SYN), encoded by the Synuclein Alpha gene (SNCA), is primarily localized within synapses but also functions in the nucleus and is the main component of Lewy bodies in Parkinson’s Disease (PD) [3]. The aggregation of α-SYN in Dopaminergic (DA) neurons, characteristic of PD, is accompanied by neuronal synaptic damage, including loss of synapses and diminished expression levels of synaptic-related proteins [4]. Some Short-Chain Fatty Acids (SCFAs), such as sodium butyrate (NaB), can activate the autophagy pathway, reducing rotenoneinduced α-SYN expression and promoting α-SYN degradation, which is essential for slowing PD progression [5]. This evidence supports a core role of α-SYN in PD development; consequently, targeting α-SYN represents a key therapeutic strategy for PD and related neurodegenerative disorders.
α-SYN expression is epigenetically regulated by histone modifications interacting with the SNCA promoter, such as H3K27ac, H3K4me1/3, H3K9me1/2, H3K14ac, H3K18ac, and H2AK119ub, which can increase SNCA expression and elevate α-SYN levels, thereby heightening PD risk [6-10]. Elevated α-SYN levels in DA neurons may further influence the epigenome through various mechanisms [3,8], creating a vicious cycle that ultimately results in the development of PD. In contrast, the repressive mark H3K9ac can repress SNCA expression and decrease α-SYN levels, thereby reducing PD risk [9]. These epigenetic deregulations of SNCA, leading to altered α-SYN expression, play a critical pathophysiological role in PD and other synucleinopathies [8].
Specifically, in the PD brain, H3K27ac is a highly common epigenetic mark across the genome, with a strong predilection for PD-associated genes such as SNCA, PARK7, PRKN, and MAPT [6]. SNCA overexpression, i.e., elevated α-SYN, may induce H3K27ac changes in the PD hippocampus [8]. Another important transcription-promoting mark, H3K4me3, interacts with the SNCA promoter and is significantly elevated at the SNCA promoter region in PD substantia nigra (SN), controlling α-SYN expression in DA neurons [7]. JARID1A, which can demethylate H3K4me3, can reduce α-SYN expression [7]. Additionally, H3K4 monomethylation can also alter SNCA mRNA expression and is significantly associated with excess α-SYN [8]. Furthermore, α-SYN overexpression leads to an elevation of H3K9me and H3K9me2 [3]. Two other acetylation marks, H3K14ac and H3K18ac, could lead to a net increase in histone H3 acetylation, enhancing SNCA gene transcription and α-SYN expression in the PD primary motor cortex [9]. Furthermore, H2AK119ub represses the phosphorylation of α-SYN at serine 129 (α-SYN-S129p). When H2AK119ub is repressed by hyperphosphorylated BMI-1 (epigenetic polycomb repressor complex-1 subunit) that undergoes a canonical polycomb E3 ligase function loss, α-SYN-S129p may accumulate, facilitating the development of PD [10]. Conversely, H3K9ac might repress SNCA gene transcription and α-SYN expression, thus reducing PD risk. Decreased H3K9ac has been found in the primary motor cortex of PD patients [9].
The following histone tail modifications interacting with other genes may also influence PD risk by regulating non-SNCA genes, exerting consistent or opposing effects to their impact on SNCA or α-SYN:
H3K27ac
Rotenone may bind to the CTCF binding sites in some PDassociated genes (e.g., PARK2, GPRIN3, FER), increase H3K27ac around these sites, impact higher-order chromatin organization, and enhance the influence of non-coding variants on genome integrity and cellular survival, thereby increasing PD risk [11], consistent with the role of α-SYN. Conversely, the H3K27ac inhibitor GNE-049 may reduce H3K27ac deposition, decrease iNOS, increase ARG1 and IRF4 levels, reduce mitochondrial superoxide, circularity, stress, and membrane depolarization, benefiting mitochondrial function, preventing primary trigger immune memory formation, and attenuating secondary inflammatory responses, thus reducing PD risk [12]. Additionally, electromagnetic stimulation activates the histone acetyltransferase Brd2, resulting in H3K27ac acetylation, alleviating PD symptoms [13].
H3K4me3
GSK-J4, a histone demethylase inhibitor, can rescue abnormal histone H3K4me3 methylation changes, contributing to its neuroprotective effects [14]. Acting as a potent iron suppressor, GSK-J4 significantly and selectively reduces intracellular labile iron in dopaminergic neurons, suppresses cell death, and diminishes oxidative stress, all of which are hallmarks of PD [14]. Hence, GSK-J4’s ability to suppress iron levels, reduce oxidative stress, and provide neuroprotection underscores its therapeutic potential for PD.
H2AX
Prx5 shields against DNA damage induced by rotenone, suppresses Ataxia Telangiectasia Mutated (ATM) kinase activity, decreases phosphorylated histone H2AX (γ-H2AX) levels, and guards against rotenone-induced neurodegeneration of dopaminergic (DA) neurons, thus mitigating the risk of PD [15]. Moreover, the expression of H2AX (Y142F) confers increased resistance to DNA damage and consequent cell death in DA neuron-like cells. This variant enhances the resilience of dopamine neurons derived from bone marrow mesenchymal stem cells (BMSCs) in a neurotoxic milieu, providing protection against the risk of PD [16].
H3K9ac
H3K9ac exhibits contrasting effects in the regulation of p53 and PGC-1α genes compared to SNCA [5,17]. SIRT1 can reduce H3K9ac levels, suppressing p53 gene transcription, boosting neuroprotection, and alleviating rotenone-induced degeneration of dopaminergic neurons, consequently lowering the risk of PD [17]. Moreover, NaB can enhance H3K9ac levels at the PGC-1α promoter, safeguarding against rotenone-induced toxicity by activating the autophagy pathway through increased expression of PGC-1α, thereby decelerating PD progression [5]. Conversely, inhibited H3K9ac may lead to the downregulation of transcription of autophagy-related genes LC3 and Atg5 [18].
H3K9me3
SIRT1, a deacetylase enzyme, can bind to H3K9 at the p53 promoter, resulting in increased H3K9me3 levels, which inhibit p53 gene transcription, enhance neuroprotection, alleviate rotenone-induced dopaminergic neurodegeneration, and thereby reduce the risk of PD or offer a novel treatment strategy for the condition [17]. Notably, H3K9me3 plays a role opposite to H3K9me and H3K9me2 on α-SYN. Neural stem/precursor cells (NSCs) represent a promising systemic cell source for PD therapy. Vitamin C (VC) has the potential to eliminate the repressive histone code H3K9me3 at specific gene promoter regions, thereby promoting the generation of authentic midbrain-type Dopamine (mDA) neurons and enhancing survival and function of NSCs derived from the Ventral Midbrain (VM). Consequently, transplantation of VC-treated NSCs could significantly enhance behavioral restoration and DA neuron engraftment, offering a straightforward, effective, and safe therapeutic avenue for PD [19]. In this context, H3K9me3 plays a role consistent with H3K9me and H3K9me2 on α-SYN.
H3K27me3
H3K27me3 functions as a repressive mark, and the histone H3K27me3 demethylase Jumonji domain-containing 3 (Jmjd3) may inhibit H3K27me3 in the SNAI2 promoter, leading to the upregulation of SNAI2 expression and activation of the SNAI2/ YAP/HIF1α signaling pathway, ultimately diminishing cell apoptosis and preserving dopaminergic neuron survival, thereby exerting a protective effect on PD [20]. Targeting the JMJD3-SNAI2 pathway holds promise as a therapeutic strategy for PD. Additionally, Jmjd3 may modify H3K27me3 to enhance the polarization of M2 microglia, contributing to the immune pathogenesis of PD. Suppression of Jmjd3 in the Substantia Nigra (SN) may elevate H3K27me3 levels, leading to microglial overactivation and exacerbating dopamine (DA) neuron death in PD. Furthermore, suppression of Jmjd3 in N9 microglia and midbrain, accompanied by an elevated level of H3K27me3, may result in extensive neuron death in PD as well [21]. The polycomb repressive complex 2 catalyzes polycomb group (PcG) proteins to bind to and repress genes in embryonic stem cells, characterized by the presence of H3K27me3 [22]. Administration of the dopamine precursor, L-DOPA, in PD induces a notable increase in H3K27me3S28 phosphorylation in medium spiny neurons expressing dopamine D1 receptors. This phosphorylation may upregulate a subset of PcG-repressed genes, contributing to longterm maladaptive responses, including motor complications or dyskinesia in PD [22]. Transplantation of VC-treated NSCs may harness their therapeutic potential for PD via H3K27me3 [22], akin to H3K9me3 as discussed above, while GSK-J4 may exert its therapeutic potential for PD via H3K27me3 [14], similar to H3K4me3 as discussed earlier.
Conclusion and future directions
Evidence has demonstrated that histone tail modifications play critical roles in the risk, development, progress, and severity of PD. These modifications could be potential pharmaceutical targets of the treatment of PD.
Histone tail modifications engage in complex interplay, or crosstalk, forming intricate regulatory networks essential for understanding the dynamic regulation of gene expression, which is a significant future research direction. These modifications are tightly regulated by specific enzymes, known as writer and reader proteins, such as histone methyltransferases, demethylases, acetyltransferases, and deacetylases. Reader proteins recognize and bind to specific modifications, recruiting other effector molecules to modulate gene expression, making the study of these enzymes’ effects another key research direction. Analyzing these enzymes’ roles and regulatory networks in the context of PD is crucial for a comprehensive understanding of histone modification regulation. Additionally, future research should focus on identifying specific histone modifications or regulatory mechanisms and exploring how these modifications can be targeted to develop novel therapeutic strategies for PD.
References
- Deliz JR, Tanner CM, Gonzalez-Latapi P. Epidemiology of Parkinson’s Disease: An Update. Curr Neurol Neurosci Rep. 2024; 24(6): 163-179.
- Bendandi A, Patelli AS, Diaspro A, Rocchia W. The role of histone tails in nucleosome stability: An electrostatic perspective. Comput Struct Biotechnol J. 2020; 18: 2799-2809.
- Sugeno N, Jackel S, Voigt A, Wassouf Z, Schulze-Hentrich J, et al. alpha-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Scientific reports. 2016; 6: 36328.
- Zhang Z, Wang R, Zhou H, et al. Inhibition of EHMT1/2 rescues synaptic damage and motor impairment in a PD mouse model. Cellular and molecular life sciences: CMLS. 2024; 81(1): 128.
- Zhang Y, Xu S, Qian Y, et al. Sodium butyrate attenuates rotenone-induced toxicity by activation of autophagy through epigenetically regulating PGC-1alpha expression in PC12 cells. Brain research. 2022; 1776: 147749.
- Toker L, Tran GT, Sundaresan J, et al. Genome-wide histone acetylation analysis reveals altered transcriptional regulation in the Parkinson’s disease brain. Mol Neurodegener. 2021; 16(1): 31.
- Guhathakurta S, Kim J, Adams L, et al. Targeted attenuation of elevated histone marks at SNCA alleviates alpha-synuclein in Parkinson’s disease. EMBO Mol Med. 2021; 13(2): e12188.
- Schaffner SL, Wassouf Z, Hentrich T, Nuesch-Germano M, Kobor MS, et al. Distinct impacts of alpha-synuclein overexpression on the hippocampal epigenome of mice in standard and enriched environments. Neurobiol Dis. 2023; 186: 106274.
- Gebremedhin KG, Rademacher DJ. Histone H3 acetylation in the postmortem Parkinson’s disease primary motor cortex. Neuroscience letters. 2016; 627: 121-125.
- Srivastava AK, Choudhury SR, Karmakar S. Neuronal Bmi-1 is critical for melatonin induced ubiquitination and proteasomal degradation of alpha-synuclein in experimental Parkinson’s disease models. Neuropharmacology. 2021; 194: 108372.
- Freeman DM, Wang Z. Epigenetic Vulnerability of Insulator CTCF Motifs at Parkinson’s Disease-Associated Genes in Response to Neurotoxicant Rotenone. Frontiers in genetics. 2020; 11: 627.
- Huang M, Malovic E, Ealy A, et al. Microglial immune regulation by epigenetic reprogramming through histone H3K27 acetylation in neuroinflammation. Front Immunol. 2023; 14: 1052925.
- Yoo J, Lee E, Kim HY, et al. Electromagnetized gold nanoparticles mediate direct lineage reprogramming into induced dopamine neurons in vivo for Parkinson’s disease therapy. Nat Nanotechnol. 2017; 12(10): 1006-1014.
- Mu MD, Qian ZM, Yang SX, Rong KL, Yung WH, et al. Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 2020; 11(10): 927.
- Wang MJ, Huang HY, Chiu TL, Chang HF, Wu HR. Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis. Cells. 2019; 9(1).
- Jiang P, Huang P, Yen SH, Zubair AC, Dickson DW. Genetic modification of H2AX renders mesenchymal stromal cell-derived dopamine neurons more resistant to DNA damage and subsequent apoptosis. Cytotherapy. 2016; 18(12): 1483-1492.
- Feng Y, Liu T, Dong SY, et al. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. Journal of neurochemistry. 2015; 134(4): 668-676.
- Zhu J, Xu F, Lai H, et al. ACO2 deficiency increases vulnerability to Parkinson’s disease via dysregulating mitochondrial function and histone acetylation-mediated transcription of autophagy genes. Commun Biol. 2023; 6(1): 1201.
- Wulansari N, Kim EH, Sulistio YA, Rhee YH, Song JJ, et al. Vitamin C-Induced Epigenetic Modifications in Donor NSCs Establish Midbrain Marker Expressions Critical for Cell-Based Therapy in Parkinson’s Disease. Stem Cell Reports. 2017; 9(4): 1192-1206.
- Dong L, Gao L. JMJD, SNAI. Synergistically protect against Parkinson’s disease by mediating the YAP/HIF1alpha signaling pathway in a mouse model. Hum Mol Genet. 2023; 32(21): 3040-3052.
- Tang Y, Li T, Li J, et al. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014; 21(3): 369-380.
- Sodersten E, Feyder M, Lerdrup M, et al. Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental parkinsonism. PLoS Genet. 2014; 10(9): e1004574.