
SciBase Journals
SciBase Obstetrics and Gynecology
ISSN 2691-7785
- Article Type: Case Report
- Volume 2, Issue 1
- Received: Nov 17, 2023
- Accepted: Feb 08, 2024
- Published Online: Feb 15, 2024
Navigating the complexities of partial androgen insensitivity syndrome: A comprehensive case study
Govani DJ1; Zaparackaite I2; Singh SJ3; Bhattacharya D4; Swamy KB5 ; Correia RC6; Midha PK7; Patel RV8*
1Medical Student, B J Med College and New Civil Hosp, Ahmedabad, Gujarat, India.
2Dept of Ped Surgery, Emergency’s Children’s Surg Hosp, Entebbe/ Evelina Children’s Hosp, London.
3Department of Pediatric Surgery, Nottingham University Hospitals, Nottingham, UK.
4Department of Pediatric Surgery, Sultan Qaboos Hospital, Salalah, Sultanate of Oman.
5Lincoln University College, Lincoln University, Kuala Lumpur, Malaysia.
6Santa Casa de Tatui Gen Hospital, 330 Maneco Pereira Road, Tatui 18273000, Sao Paulo, Brazil.
7J. Watumull Global Hospital & Research Centre, Delwara Road, Mount Abu, Rajasthan, India affiliated to Medical Faculty of God Fatherly World Spiritual University, Mount Abu, Rajasthan.
8Departments of Pediatrics and Ped Surgery, Postgraduate Institute of Child Health & Research and KT Children Govt University Teaching Hospital, Rajkot 360001, Gujarat, India.
*Corresponding Author: Ramnik V Patel
Department of Pediatric Surgery, Postgraduate Institute of
Child Health and Research and K T Children Government
University Teaching Hospital, Rajkot 360005, Gujarat, India.
Tel: 00447956896641;
Email: ramnik@doctors.org.uk
Abstract
Partial Androgen Insensitivity Syndrome (PAIS) is a rare genetic disorder that poses unique challenges in clinical management. This case study delves into the clinical presentation, diagnostic journey, and management of a patient diagnosed with PAIS. The complexities of androgen receptor dysfunction and its impact on sexual development are explored, providing valuable insights for clinicians and researchers alike.
Keywords: Partial androgen insensitivity syndrome; Androgen receptor dysfunction; Sexual development; Clinical management; Genetic disorder.
Citation: Govani DJ, Zaparackaite I, Singh SJ, Bhattacharya D, Patel RV, et al. Navigating the complexities of partial androgen insensitivity syndrome: A comprehensive case study. SciBase Obstet Gynecol. 2024; 2(1): 1004.
Introduction
Androgen receptor sensitivity is very crucial factor not only in the developing fetus but at puberty to allow full expression of male phenotype in an XY genotype person [1]. The androgen insensitivity syndrome (AIS) represents a spectrum of disorders where the degree of receptor insensitivity varies from minimal to complete insensitivity [2]. Partial Androgen Insensitivity Syndrome (PAIS) is a rare X-linked genetic disorder characterized by impaired androgen receptor function, leading to a spectrum of phenotypic variations in individuals with a 46, XY karyotype. This case study aims to illuminate the clinical manifestations, diagnostic challenges, and therapeutic considerations associated with PAIS, contributing to a deeper understanding of this complex condition.
Case report
A 25-year-old married apparently female patient with a poor rural background, was brought to us for investigation of primary infertility following 2 years of their married life. She was first in order of birth and had noticed some weight gain recently. A detailed history revealed that she was born with apparent female genitalia, had poor breast development, no menarche and had never had menstrual periods.
On examination, patient had average height and weight. There were no overt signs of masculinisation but had scanty axillary and pubic hair, and poor breast development to Tanner’s stage III. Abdominal examination was normal. No gonads or hernia can be seen or felt in the labioscrotal fold or groin. Perineal examination showed slightly prominent clitoris with three distinct openings of urethra, vagina and anal openings. However, there was no remnants of the hymen and it was shorter and blind ending without any cervix or palpable uterus. Rectal examination could not feel uterus. Sexual developmental disorder was suspected.
Investigations showed normal serum electrolytes and urinary 17-ketosteroids. Hormone profile revealed normal serum dehydroepiandrosterone sulphate, androstenedione, and 17-hydroxyprogesterone levels. But had high serum testosterone levels, low serum oestradiol and elevated Serum LH and serum FSH. Ultrasound scan of the abdomen and pelvis revealed a marked hypoplastic uterus measuring 3x1x5 cm with no evidence of any intra-abdominal gonads. Rest of the abdominal and pelvic structures were normal (Figure 1).
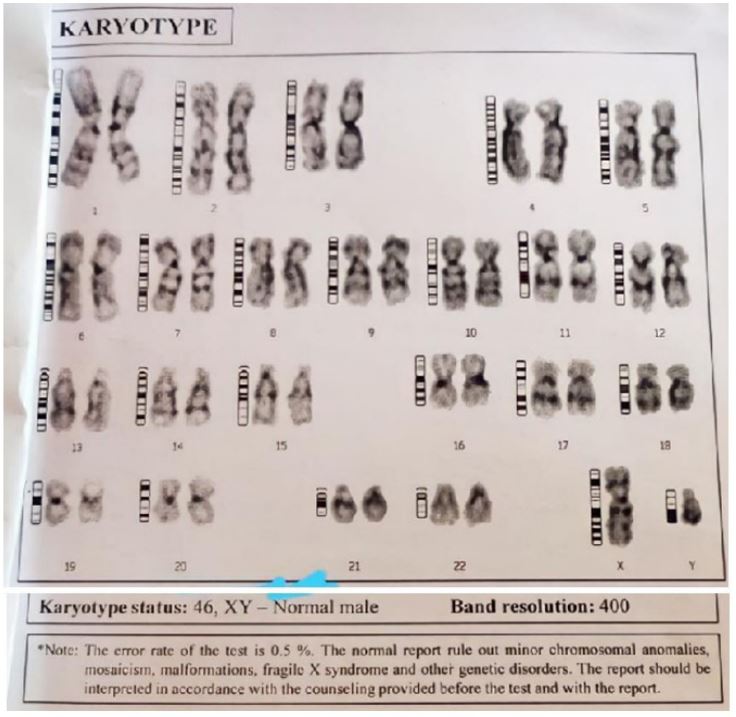
Barr body was present on the buccal mucosal smear. The karyotype on 5 metaphase cells revealed 46 XY typical male pattern (Figure 2). There were no numerical or structural chromosomal anomalies. In the clinical setting it was reported as partial testicular feminisation (Figure 3).
Multidisciplinary meeting of disorders of sexual differentiation was held. Patient underwent diagnostic laparoscopy at which bilateral gonads with rudimentary fallopian tubes connected to the rudimentary uterus were seen. Bilateral gonadectomy with removal of rudimentary Mullerian remnants was performed uneventfully. Histopathological examination confirmed the gonads to be testis and the tubules to be fallopian tubes. A final diagnosis of PAIS was made.
The endocrine team prescribed hormone replacement therapy with cyclic oestrogen and progesterone for the breast development. Genetic and psychological counselling was carried out. For the fertility options surrogate pregnancy with an oocyte donor having baby with three mothers for which the couple is actively searching the oocyte donor and surrogate mother failing which may accept adoption or accept the situation as it is.


Discussion
In our culture, it is very usual to find delay in seeking medical help due to lack of education and especially in poor rural community due to not being aware about personal hygiene and health education. Moreover, there is distinct shyness when it comes to sex related matters. Androgen insensitivity syndrome (AIS) spectrum can manifest in three forms; minimal AIS (MAIS) the patient is a phenotypically male with male sterility. gynaecomastia, and azoospermia, in the middle of the spectrum is PAIS that is the most challenging to diagnose early and presents as a diagnostic dilemma while the other end of the spectrum is complete AIS (CAIS) who are XY genotypically and are usually tall phenotypically females with well-developed breasts, blind vagina, and absent or scanty pubic and axillary hair [3,4].
Partial androgen insensitivity syndrome (PAIS) is an X-linked recessive disorder characterized by incomplete male sexual development and variable degrees of androgen insensitivity. The disorder results from mutations in the androgen receptor (AR) gene, leading to impaired androgen signalling and ultimately the clinical manifestations of PAIS. The prevalence of PAIS is estimated to be 1 in 20,000 to 1 in 64,000 males, while the incidence of PAIS is one in 130,000 births [5]. The estimated incidence of CAIS is one in 20,400 XY births [6].
Genetic base of PAIS is on AR gene. The AR gene is located on the X chromosome and encodes the AR protein. The AR plays a crucial role in male sexual development and function by mediating the effects of androgens, such as testosterone and dihydrotestosterone (DHT). Mutations in the AR gene can disrupt the AR’s ability to bind androgens, translocate to the nucleus, and activate androgen-responsive genes, leading to impaired androgen signalling and the clinical manifestations of PAIS.
There are 2 types of AR mutations:
1. Ligand-binding domain (LBD) mutations: These mutations disrupt the AR’s ability to bind androgens, such as testosterone and DHT.
2. DNA-binding domain (DBD) mutations: These mutations disrupt the AR’s ability to bind to androgen-response elements (AREs) in the promoter regions of target genes.
AR mutations in PAIS lead to a spectrum of androgen signalling defects, ranging from complete androgen insensitivity to mild androgen resistance. The severity of the androgen signalling defect depends on the type and location of the AR mutation [7].
The clinical manifestations of PAIS vary depending on the severity of the androgen signalling defect. In general, individuals with PAIS may have the following ambiguous genitalia: Individuals with PAIS may have ambiguous genitalia, cryptorchidism, gynecomastia, primary amenorrhea and infertility due to impaired spermatogenesis.
Diagnosis of PAIS is based on a combination of clinical presentation, physical examination, laboratory tests, imaging and diagnostic endoscopy and biopsies. Genetic testing can confirm the diagnosis and identify the specific AR mutation present in the individual.
Treatment options for PAIS depend on the severity of the androgen signalling defect and the individual’s specific needs.
Treatment may include:
1. Hormone replacement therapy: Hormone therapy with testosterone or dihydrotestosterone (DHT) can help masculinize the body and promote male secondary sex characteristics. Female phenotypes may require cyclical estrogen and progesterone for breast development as in our case.
2. Surgery: Surgery may be necessary to correct ambiguous genitalia with genitoplasty or undescended testis with gonadectomy in relation to tumour risk.
3. Psychological counselling: Gender assignment at birth is a psycho-social emergency. Psychological counselling can help individuals with PAIS cope with the emotional and social challenges of the disorder.
4. Genetic Counselling for PAIS is recommended for individuals with PAIS and their families. Genetic counselling can provide information about the disorder, its genetic basis, and the risks of recurrence in future pregnancies. Genetic testing can confirm the diagnosis of PAIS and identify the specific AR mutation present in the individual. This information can be helpful in guiding treatment decisions and providing reproductive counselling.
Gonads may be observed in CAIS cases with close observation and supervision [3,4]; while for PAIS presenting with undescended testes, gonadectomy at time of diagnosis followed by hormone replacement therapy is currently recommended due to the high (50%) risk of germ cell malignancy [8].
Conclusion
PAIS is a complex disorder with a wide range of clinical manifestations. Understanding the genetic basis of PAIS and the impact of AR mutations on androgen signalling is crucial for providing accurate diagnosis, appropriate treatment, and informed genetic counselling. Most cases of PAIS should be raised as males but if, as in our case reared as female being on the milder form of the spectrum, a multidisciplinary approach with tailor made decision is essential to reach the best prognosis for satisfaction of patients/parents, professionals and public.
References
- Mohan S, Kapoor G, Raman DK. Partial androgen insensitivity syndrome: a diagnostic and therapeutic dilemma. Med J Armed Forces India. 2011; 67(4): 382-4. doi: 10.1016/S0377-1237(11)60093-2. Epub 2011 Oct 22. PMID: 27365856; PMCID: PMC4920682.
- Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A. Androgen insensitivity syndrome: clinical features and molecular defects. Hormones (Athens). 2008; 7: 217-229.
- Patel RV, Govani ND, Govani DR, Panchasara NG, Patel RR, Midha PK. Oocyte Donation and Surrogate Pregnancy-Fertility Options and Challenges, Possibilities and Opportunities for a Patient with Complete Androgen Insensitivity Syndrome. Medp J Obstet Gynaecol. 2022; 1(1): 202203001.
- Patel RV, Govani ND, Govani DR, Panchasara NG, Patel RR, Midha PK, A Case of Familial Complete Androgen Insensitivity Syndrome- Baby with Three Mothers and the Daughter shows the mother—Challenges and Threats in Advanced Assisted Reproduction Technology. Medp Pediatr Child Health Care. 2022; 1(1): 202203002.
- Bangsbøll S, Qvist I, Lebech PE, Lewinsky M. Testicular feminization syndrome and associated gonadal tumors in Denmark. Acta Obstet Gynecol Scand. 1992; 71: 63-66.
- Lund A, Juvonen V, Lähdetie J, Aittomäki K, Tapanainen JS, Savontaus ML. A novel sequence variation in the transactivation regulating domain of the androgen receptor in two infertile Finnish men. Fertil Steril. 2003; 79(3): 1647-1648.
- Hughes IA, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab. 2006; 20: 577-598.
- Ahmed SF, Cheng A, Dovey L. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000; 85: 658-665.